As you know, in deciding which position undergoes aromatic electrophilic substitution, three elements must be taken in account: steric effect, mesomeric effect and inductive effect.
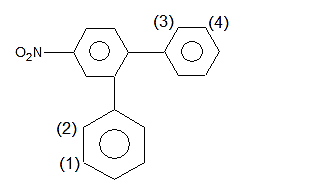
Let's call the ring containing atoms 1 and 2 "A", and the one containing atoms 3 and 4 "B".
Before deciding the importance of any of those effects, some considerations about the geometry of the molecule should be taken in account. It could be guessed, by thinking about the spatial arrangement of such molecule, that the high steric effect between rings A and B prevents their rotation, thus keeping them in a constantly "skewed" position in respect to the nitrated ring.
A representation of the molecule, with the Van Der Waals radius of the elements, follows:
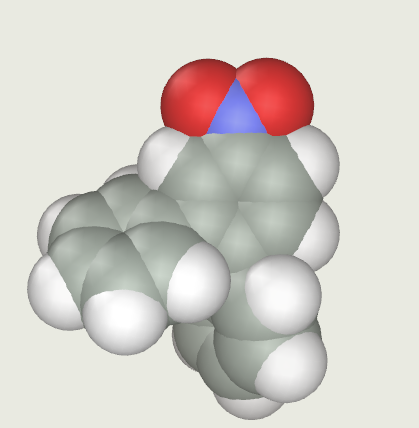
Steric Effects: Because of the presence of a nearby phenyl (the nitrated one) to any of the ortho positions of rings A and B, and because of the presence of the other phenyl in the middle of the possible electrophile's path, all the ortho positions might be considered as non favourable.
Mesomeric effect: One might guess that the resonance structures of the nitrated ring could extend to ring B, while leaving ring A untouched.
Actually, such a resonance structure requires a "double bond behaviour" for the aromatic carbons between ring B and the nitrated ring. But the molecule is extremely unlikely to behave that way, since the copresence of two aromatic rings prevents their own rotation, and locks them in a skewed position. This conformation, in other words, renders the pi system "less conjugated", and the mesomeric effect much less relevant.
Inductive effect: This is the final and, in this specific molecule, most important consideration. Simply, everything boils down to the proximity of the nitro EWG: this makes ring A more electron-poor, and ring B more electron-rich.
The answer is, hence: the meta position of ring B is attacked, or the "4" position in your image.