tl;dr: I don't think there is any mechanism that is 100% correct, and, in cases like this especially, I think it would completely depend upon what set of carbonyl/ylid/base/solvent etc. was used. But, of course, we like being able to generalise, and to my knowledge theres a lot more evidence to support a concerted type mechanism.
General Background
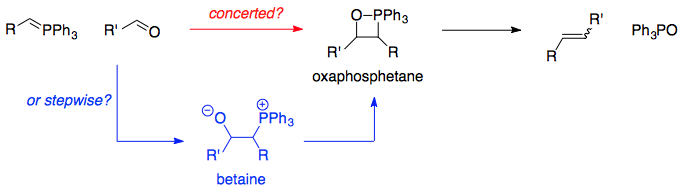
The question is hopefully summarised in the scheme below (no geometry, that, in itself, is a pretty lengthy discussion). Reaction of a carbonyl (in this case a ketone) with a phosphorus ylid is able to give rise to two species, either an oxaphosphetane directly or a betaine (an internal salt for the benefit of Loong) which then goes on to form the oxaphosphetane which itself is able to eliminate to afford the olefin with generation of triphenylphosphine oxide as a thermodynamic driving force.
This answer will present the case for a cycloaddition mechanism, and evidence against the betaine pathway. Importantly, only Wittig reactions of unstabilised ylids, as with stabilised ylids the rules of the game change due to reversibility of intermediate formation (see a Horner–Wadsworth–Emmons).
Evidence against the initial formation of a betaine
Betaines have never been observed during the course of a 'normal' Wittig reaction, that is, spectroscopically, we are unable to see it (this may, of course, just be due to the fact that the formation of the oxaphosphetane is so rapid relative to the timescale of the methods used).
Many reports of betaines have been from accidental (or otherwise) opening of oxaphosphetane intermediates, and indeed this seems to have been what led Wittig down the betaine pathway in the initial reports (though in the first ever paper he suggested that oxaphosphetane was the sole intermediate, but subsequently couldn't find any evidence for it back in the days before NMR etc).
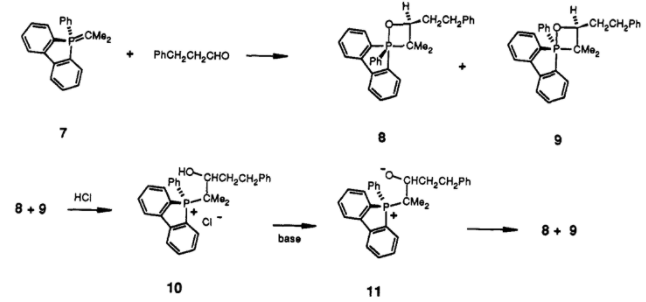
Wittig[2] and others[3] reported the isolation of crystalline betaine salts. Phosphonium bromides were treated with PhLi to give a ylid, to which a carbonyl was added; subsequent addition of hydrogen bromide afforded crystalline solids which were able to be elucidated, proving the formation of a betaine.
Whilst this was convincing at the time, we now know that the betaine isn't observed, but rather the oxaphosphetane is. Vedejs (whom we'll come back to), has gone on to show that the earlier findings could be explained by quenching of the oxaphosphetane rather than as a result of directly trapping the betaine[4], which is more consistent with the other data available to us.
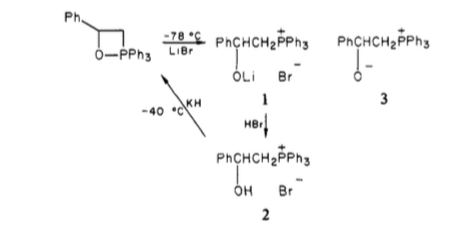
Scheme 1: Generation of the crystalline betaine derivative from the oxaphosphetane.
There are more recent reports of 'stable' betaines for very specific substrates where subsequent collapse isn't possible, again generated from oxaphosphetane intermediates.
Stefan Berger's group at Leipzig reported a betaine stabilised by a bipyridyl group, allowing NMR data to be reported for the first time[1].
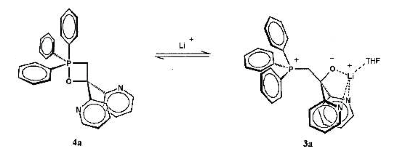
Scheme 2 Reversible formation of a stabilised betaine
In this case, the lithium holds a chelated structure together, preventing it from forming the alkene (and also to a certain extent preventing reformation of the oxaphosphetane on steric grounds). Interesting, with addition of 18-crown-6 to sequester the lithium, the reaction proceeded normally in the forward direction with alkene signals observed.
The fact Berger was able to do this, does of course not imply that they are real intermediates along the Wittig pathway, but it was a nice piece of mechanistic work that I thought deserved mention.
In addition to this mechanistic work, there are some empirical issues with invoking a betaine intermediate that cannot be easily explained. Namely, all Wittig reactions using non-stabilised ylids should be under kinetic control and irreversible (hence highly (Z)-selective due to the inability of the initial intermediates to reverse and hence equilibrate to the thermodynamic product), however this is frequently observed not to be the case, and in certain cases, high (E)-selectivity can be achieved.
Evidence for direct oxaphosphetane formation
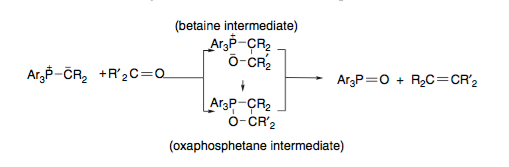
Vedejs was the one of the first to propose direct (irreversible) cycloaddition of the ylid and carbonyl to give rise to the oxaphosphetane, quickly followed by cycloreversion to form the desired alkene and a phosphine oxide.
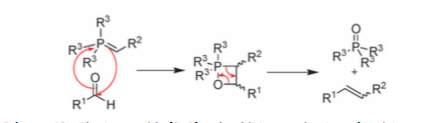
Scheme 3: cycloaddition/cycloreversion mechanism for Wittig
In his early reports, direct observation of the oxaphosphetane by P-NMR was conducted[5], the paper reasons that the high-field chemical shift observed for the phosphorus containing intermediate ruled out charged, 4-valent species such as betaines, but was instead more consistent with formation of the 5-valent neutral species such as an oxaphosphetane.

Scheme 4: Vedejs' observation of oxaphosphetane.
This NMR analysis isn't really too convincing by itself, however in the scheme above, compound 4 had previously been characterised by X-ray crystallography[6] and various NMR methods and as such gave some weight to the species observed by Vedejs being similar to the known compound. More recent work[7] has also observed directly the cis/trans oxaphosphetanes formed via competing cycloadditions.
Summary
Overall, direct observation of the oxaphosphetane but not the betaine does seem to suggest that mechanistically the cyclic intermediate is formed directly rather than via the betaine closing.
As stated previously, we can't 100% rule out the possibility that the betaine closure is just sufficiently rapid to appear invisible to our currently spectroscopic methods however every report of isolated/observed betaines that I've came across thus far has either intentionally been formed from an oxaphosphetane or can be explained by this without the authors recognising it.
One thing that is hugely missing from the story is computational work, whilst some studies have calculated the relative energies of the two intermediates, to my knowledge, the entire reaction coordinate hasn't been fully explored.
In conclusion, I think the cycloaddition mechanism is safest, and indeed it was the one I was taught as an undergraduate (and the one I know several other leading universities use), so if in doubt i'd go for that explanation, but on the firm understanding that more mechanistic work is needed.
References
Generally: Modern Carbonyl Olefination, Carey Advanced Organic A, and Comprehensive Organic Synthesis I.
[1] Neumann, R. A.; Berger, S. Observation of a Betaine Lithium Salt Adduct During the Course of a Wittig Reaction. Eur. J. Org. Chem. 1998, 6, 1085. Subramanyam had reported similar chemistry earlier, but the NMR data was incomplete.
[2] Wittig, G.; Haag, A. Über Phosphin-alkylene als olefinbildende Reagenzien, VIII. Allenderivate aus Ketenen. Chem. Ber. 1963, 96 , 1535. DOI: 10.1002/cber.19630960609. A very old paper in German.
[3] Schlosser, M.; Christmann, K. F. Olefinierungen mit Phosphor-Yliden, I. Mechanismus und Stereochemie der Wittig-Reaktion. Liebigs Ann. Chem. 1967, 708, 1. DOI: 10.1002/jlac.19677080102.
[4] Vedejs, E.; Meier, G. P.; Snoble, K. A. J. Low-temperature characterization of the intermediates in the Wittig reaction. J. Am. Chem. Soc. 1981, 103, 2823. DOI: 10.1021/ja00400a055.
[5] Vedejs, E.; Snoble, K. A. J. Direct observation of oxaphosphetanes from typical Wittig reactions. J. Am. Chem. Soc. 1973, 95, 5778. DOI: 10.1021/ja00798a066.
[6] Mazhar-Ul-Haque; Caughlan, C. N.; Ramirez, F.; Pilot, J. F.; Smith, C. P. Crystal and molecular structure of a four-membered cyclic oxyphosphorane with pentavalent phosphorus, PO2(C6H5)2(CF3)4C3H2. J. Am. Chem. Soc. 1971, 93, 5229. DOI: 10.1021/ja00749a044.
[7] Maryanoff, B. E.; Reitz, A. B.; Mutter, M. S.; Whittle, R. R.; Olofson, R. A. Stereochemistry and mechanism of the Wittig reaction. Diasteromeric reaction intermediates and analysis of the reaction course. J. Am. Chem. Soc. 1986, 108, 7664. DOI: 10.1021/ja00284a034.