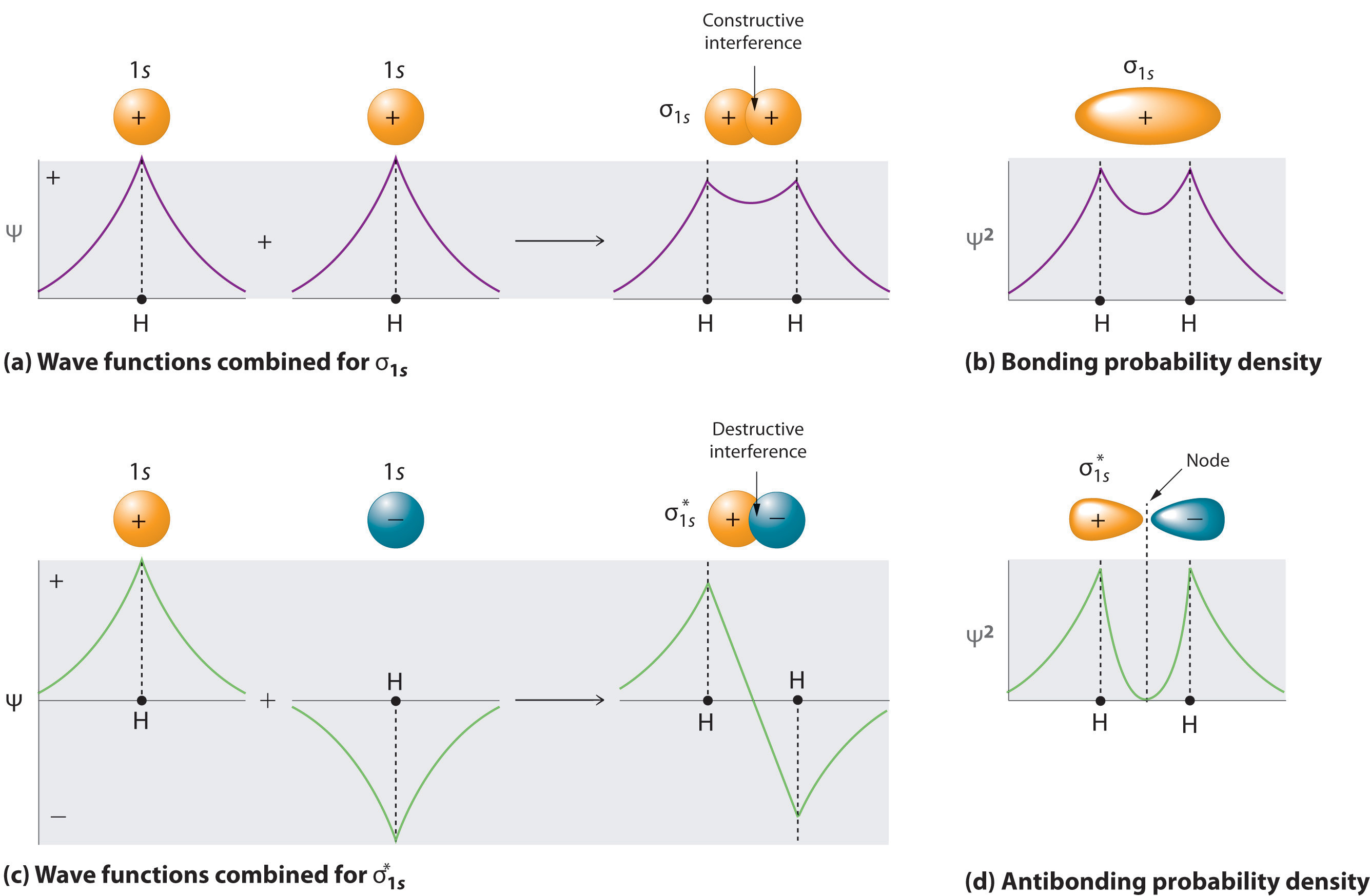
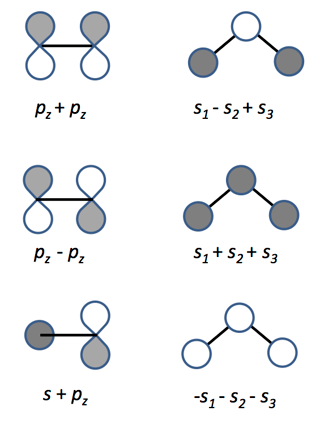In addition to @Nanoputian's excellent description of constructive and destructive interference in the formation of MOs, I want to provide a more mathematical explanation for why the phase of the wavefunction does not matter.
Finding the wavefunction
The time-independent Schrödinger equation, in one dimension, reads:
$$\hat{H}\psi(x) = E\psi(x)$$
It can be shown that, if a wavefunction $\psi = \psi(x)$ satisfies the above equation, the wavefunction $k\psi$ (with $k \in \mathbb{C}$) also satisfies the above equation with the same energy eigenvalue $E$. This is because of the linearity of the Hamiltonian:
$$\begin{align}
\hat{H}(k\psi) &= k(\hat{H}\psi) \\
&= k(E\psi) \\
&= E(k\psi)
\end{align}$$
There are several conditions that a wavefunction must satisfy for it to be physically realisable, i.e. for it to represent a "real" physical particle. In this discussion, the relevant condition is that the wavefunction must be square-integrable (or normalisable). In mathematical terms:
$$\langle\psi\lvert\psi\rangle = \int_{-\infty}^{\infty}\!\lvert\psi\rvert^2\,\mathrm{d}x < \infty$$
This means that there has to exist a constant $N \in \mathbb{C}$ such that $N\psi$ is normalised:
$$\int_{-\infty}^{\infty}\!\lvert N\psi\rvert^2\,\mathrm{d}x = \lvert N \rvert^2 \!\!\int_{-\infty}^{\infty}\!\lvert\psi\rvert^2\,\mathrm{d}x = 1$$
From this point onwards, we will assume that we will already have found the suitable normalisation constant such that the wavefunction $\psi$ is already normalised. In other words, let's assume $\langle\psi\lvert\psi\rangle = 1$, because we can. Now let's consider the wavefunction $-\psi$, which is equivalent to $N\psi$ with $N = -1$. Is this new wavefunction normalised?
$$\begin{align}
\int_{-\infty}^{\infty}\!\lvert -\psi\rvert^2\,\mathrm{d}x &= \lvert -1 \rvert^2 \!\!\int_{-\infty}^{\infty}\!\lvert\psi\rvert^2\,\mathrm{d}x \\
&= \int_{-\infty}^{\infty}\!\lvert\psi\rvert^2\,\mathrm{d}x \\
&= 1
\end{align}$$
Of course it is. So, what I've written so far basically says: if $\psi$ is a normalised solution to the Schrödinger equation, so is $-\psi$.
In fact, you could go one step further. Using exactly the same working as above, you could show that if $\psi$ is a normalised solution to the Schrödinger equation, the wavefunction $(a + ib)\psi$ would also be one, as long as $a^2 + b^2 = 1$. (If you like exponentials, that's equivalent to saying $a + ib = e^{i\theta}$.) I've illustrated this idea on this diagram:
$\qquad\qquad\qquad\qquad\qquad\qquad$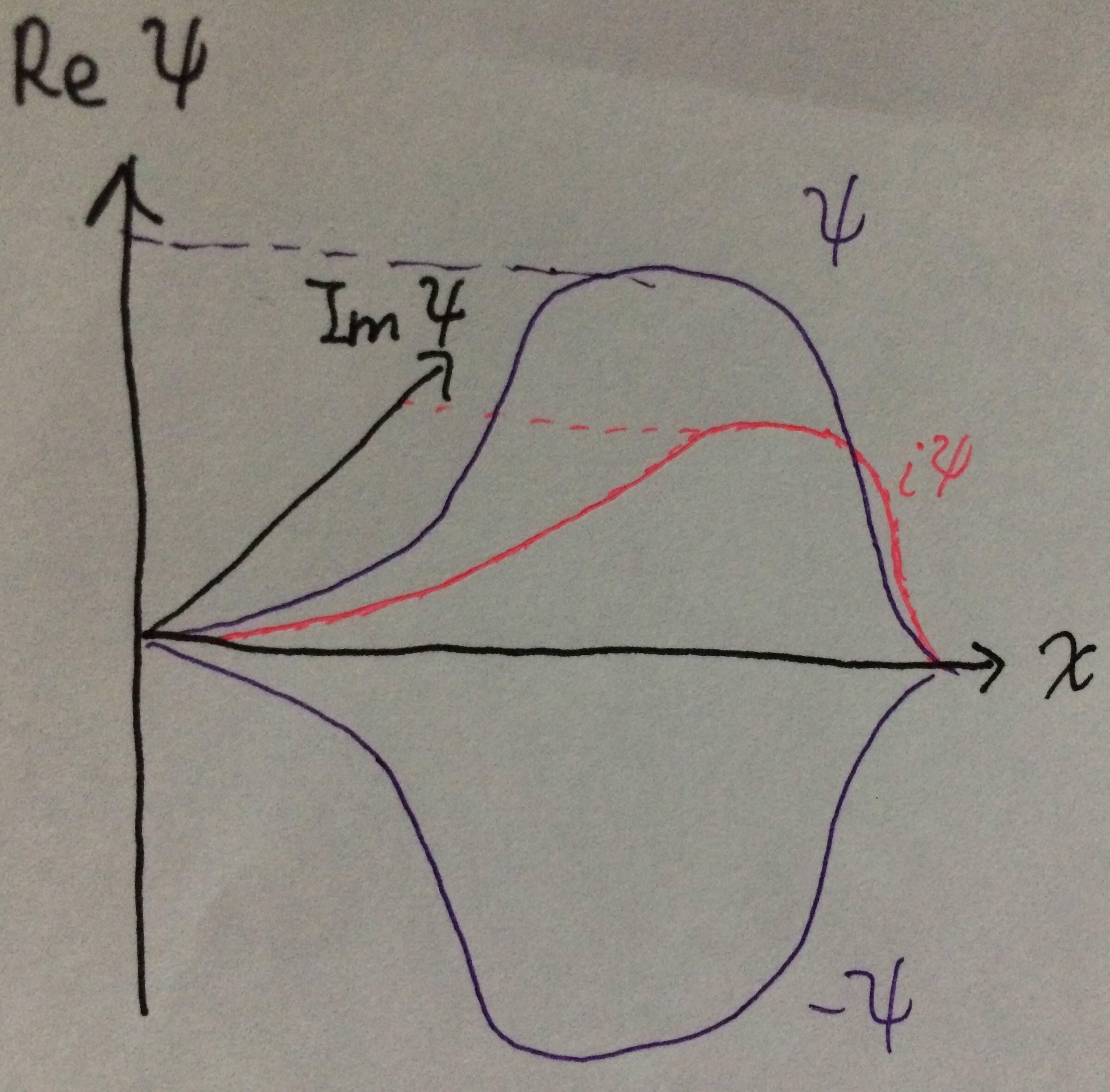
If $\psi$ is a real-valued, one-dimensional wavefunction, you could plot it on a graph against $x$. The wavefunction $i\psi$ would then be exactly the same shape, just coming out of the plane of the paper ($\theta = 90^\circ$). You could have the wavefunction $(1+i)\psi/\sqrt{2}$. It would be pointing outwards of the plane of the paper by $\theta = 45^\circ$, exactly halfway in between $\psi$ and $i\psi$, but exactly the same shape. However, physics doesn't know where the plane of your paper is, so all these wavefunctions are equally admissible. From the point of view of the system, they are all the same thing.
Using the wavefunction
"But wait! If the wavefunction is negative, what about the values of momentum, position, and energy that you calculate? Will they become negative?"
"Good question, myself!"
Well, for starters, one thing that you use the wavefunction for is to find the probability density, $P(x)$. According to Max Born's interpretation of the wavefunction, this is given by $P(x) = \lvert \psi \rvert ^2$. Let's say that the probability density described by the negative wavefunction $-\psi$ is a different function of $x$, called $Q(x)$:
$$\begin{align}
Q(x) = \lvert -\psi \rvert ^2 &= \lvert -1 \rvert^2 \lvert \psi \rvert ^2 \\
&= \lvert \psi \rvert ^2 \\
&= P(x)
\end{align}$$
So, the probability density described by the negative wavefunction is exactly the same. In fact, the probability density described by $i\psi$ is exactly the same as well.
Now let's talk about observables, such as position $x$, momentum $p$, and energy $E$. Every observable has a corresponding operator: $\hat{x}$, $\hat{p}$, and $\hat{H}$ respectively (the Hamiltonian has a special letter because it's named after William Hamilton). You use these operators to calculate the mean value of the observable. I'll give an example regarding the momentum. If you want to find the mean momentum, denoted $\langle p \rangle$, you would do the following:
$$\begin{align}
\langle p \rangle &= \langle\psi\lvert\hat{p}\rvert\psi\rangle \\
&= \int_{-\infty}^\infty\!\psi^*\hat{p}\psi\,\mathrm{d}x
\end{align}$$
I'm going to call the value of that integral $p_1$. Now, let's do the same thing. Let's assume that the mean momentum for the negative wavefunction is not necessarily the same value. Let's call the new mean momentum something else, like $p_2$.
Before we go on, I'm going to establish that the momentum operator $\hat{p} = -i\hbar\frac{\mathrm{d}}{\mathrm{d}x}$ is also linear. If you doubt it, you can test it out using the definition of linearity in the very first link I posted. In fact, all quantum mechanical operators corresponding to observables are linear. Therefore $\hat{p}(-\psi) = -\hat{p}\psi$ and so:
$$\begin{align}
p_2 &= \langle -\psi\lvert\hat{p}\lvert-\psi\rangle \\
&= \int_{-\infty}^\infty\! (-\psi)^*\hat{p} (-\psi)\,\mathrm{d}x \\
&= (-1)^2\!\!\int_{-\infty}^\infty\! \psi^*\hat{p}\psi\,\mathrm{d}x \\
&= \int_{-\infty}^\infty\! \psi^*\hat{p}\psi\,\mathrm{d}x \\
&= p_1
\end{align}$$
So, if we talk about the ground state of the particle in a box of length $L$, no matter whether you use the positive wavefunction
$$\psi_1 = \sqrt{\frac{2}{L}}\sin{\left(\frac{\pi x}{L}\right)}$$
or the negative wavefunction
$$-\psi_1 = -\sqrt{\frac{2}{L}}\sin{\left(\frac{\pi x}{L}\right)}$$
or the complex wavefunction
$$i\psi_1 = i\sqrt{\frac{2}{L}}\sin{\left(\frac{\pi x}{L}\right)}$$
you'll get exactly the same values for average position $(= L/2)$, average momentum $(= 0)$, and average energy $(= h^2/2mL^2)$ (the word average is redundant here, since this is a stationary state, but whatever).
Everything that I have said so far can be easily generalised to three dimensions. It can also be generalised to linear combinations of stationary states, i.e. solutions of the time-dependent Schrödinger equation.
A note about molecular orbitals
"Okay, but what happens when you combine atomic orbitals to make molecular orbitals? You have constructive interference from the positive + positive, and destructive interference from the positive + negative, but what about the negative + negative combination?"
"Good question, myself!"
Let's talk about the $\ce{H2}$ molecule. The proper way to find the molecular orbitals is to solve the Schrödinger equation for the entire system, which is really difficult to do. One way to find approximate forms of the MOs is to make linear combinations of atomic orbitals; this method is called the LCAO approximation. Let's call the 1s orbital of the hydrogen on the left $\phi_1$ and the 1s orbital of the hydrogen on the right $\phi_2$. From the previous sections, we have already established that as far as the hydrogen atom is concerned, the individual phases of $\phi_1$ and $\phi_2$ do not matter. So, let's assume for simplicity's sake that their phases are both positive.
Now, from what you already know, you can get two molecular orbitals $\psi_1$ and $\psi_2$:
$$\begin{align}
\psi_1 &= \phi_1 + \phi_2 \\
\psi_2 &= \phi_1 - \phi_2
\end{align}$$
These are the bonding and antibonding orbitals respectively (at least, to within a normalisation constant, which I'm not going to care about here because the details are irrelevant). Now let's talk about those combinations that we missed out.
$$\begin{align}
-\phi_1 - \phi_2 &= -\psi_1 \\
-\phi_1 + \phi_2 &= -\psi_2
\end{align}$$
We already said that $\psi_1$ and $\psi_2$ are (approximations of) solutions to the Schrödinger equation. That means that, from what we've talked about earlier, $-\psi_1$ and $-\psi_2$ must also be (approximations of) solutions to the Schrödinger equation. They must have the same energies as $\psi_1$ and $\psi_2$. In fact, as far as the molecule knows (and cares), they are the same thing as $\psi_1$ and $\psi_2$.
Now, since the individual phases of the atomic orbitals do not matter, if you really wished to, you could declare to the whole world that you define:
$$\phi_3 = \phi_1 \text{ and } \phi_4 = -\phi_2$$
i.e. left hydrogen 1s orbital, $\phi_3$, is positive and right hydrogen 1s orbital, $\phi_4$, is negative. In that case, you can construct the molecular orbitals:
$$\begin{align}
\psi_1 &= \phi_3 - \phi_4 \\
\psi_2 &= \phi_3 + \phi_4
\end{align}$$
The coefficients of the atomic orbitals would have to be different, since you insisted on having them in different phases - however, the outcome is the same! You get one bonding MO and one antibonding MO.