TLDR: Atkins' physical chemistry contains the quote "In general, from N atomic orbitals we can build N molecular orbitals". In the case of $H^+_2$, we use a combination of N=2 orbitals to create the bonding and anti-bonding orbitals as shown in eq 1. But if we allow one of the orbitals to differ by a phase $C_0$ so that $\Psi_{0}(r)=N\left[\psi_{H1s_A}(r)+C_o\psi_{H1s_B}(r)\right]$, then from two atomic orbitals we should be able to create an infinite amount of molecular orbitals. Atkins is correct. I am incorrect. But why?
TLDR Over
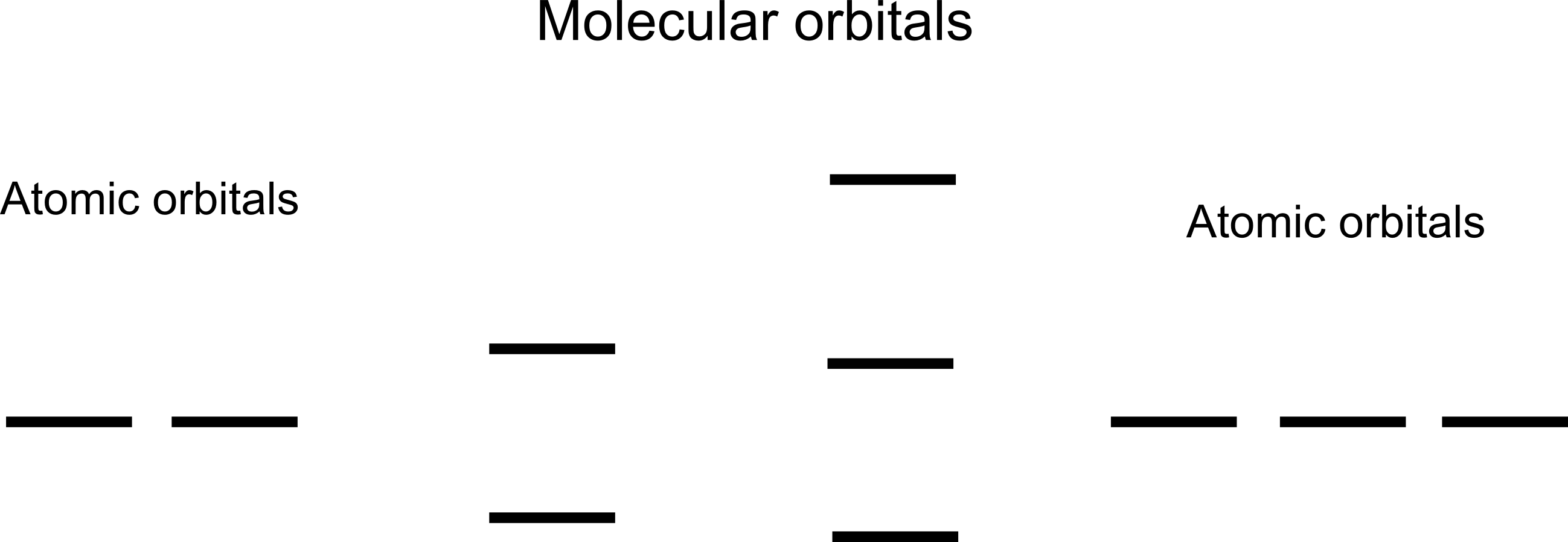
Using the LCAO method for the $H_2^+$ ion, we use as our trial functions $$\Psi_{\pm}(r)=N\left[\psi_{H1s_A}(r)\pm\psi_{H1s_B}(r)\right] \tag{1}$$ where $N$ is a normalization factor, and $\psi_{H1s_i}$ is the 1s orbital centered around the $i$'th nucleus. The wave function $\Psi_{+}$ results in a significant reduction in the systems energy whilst the $\Psi_{-}$ wavefunction significantly increases the systems energy. For this reason, $\Psi_{+}$ is called the bonding orbital whilst $\Psi_{-}$ is called the anti-bonding orbital.
This phenomenon is often described as playing a role in the origin of the band structure of solids. When N independent potential wells are brought near each other, the initially N-fold degenerate ground state "splits" into N energy eigenstates. This is simply an N-potential generalization to the $H^+_2$ case described above. In that case, we simply have $N=2$ and the $N=2$ energy eigenstates that arise as the nuclei approach each other are simply the bonding and anti-bonding states.
My issue is that I can't seem to think of any reason as to why we should restrict ourselves to only the two states $\Psi_{+}(r)$ and $\Psi_-(r)$ when performing the LCAO method. Surely the state $\Psi_{0}(r)=N\left[\psi_{H1s_A}(r)+C_o\psi_{H1s_B}(r)\right]$ (where $C_o$ is a complex number) should be just as valid as either of the $\Psi_{\pm}$ states? (Note that anti-symmetrization is not a requirement here since we are calculating 1 electron wave functions).
I also feel like depending on the value of $C_o$, the energy of this new state should vary continuously between the energy of the bonding state and the energy of the anti-bonding state? If this is the case, then the coupling that occurs when $N=2$ hydrogen nuclei are brought to within bonding distance of each other should result in an infinite continuum of energy eigenstates. Not merely two eigenstates. Similarly, when $N\approx 10^{23}$ atoms are brought within bonding distance of each other, we shouldn't get $N$ energy levels but rather an infinite amount of energy levels. This would then ruin the whole notion that energy bands in a solid of N atoms should each contain $N$ energy levels.