I'm having a hard time determining regio-selectivity in those two reactions.
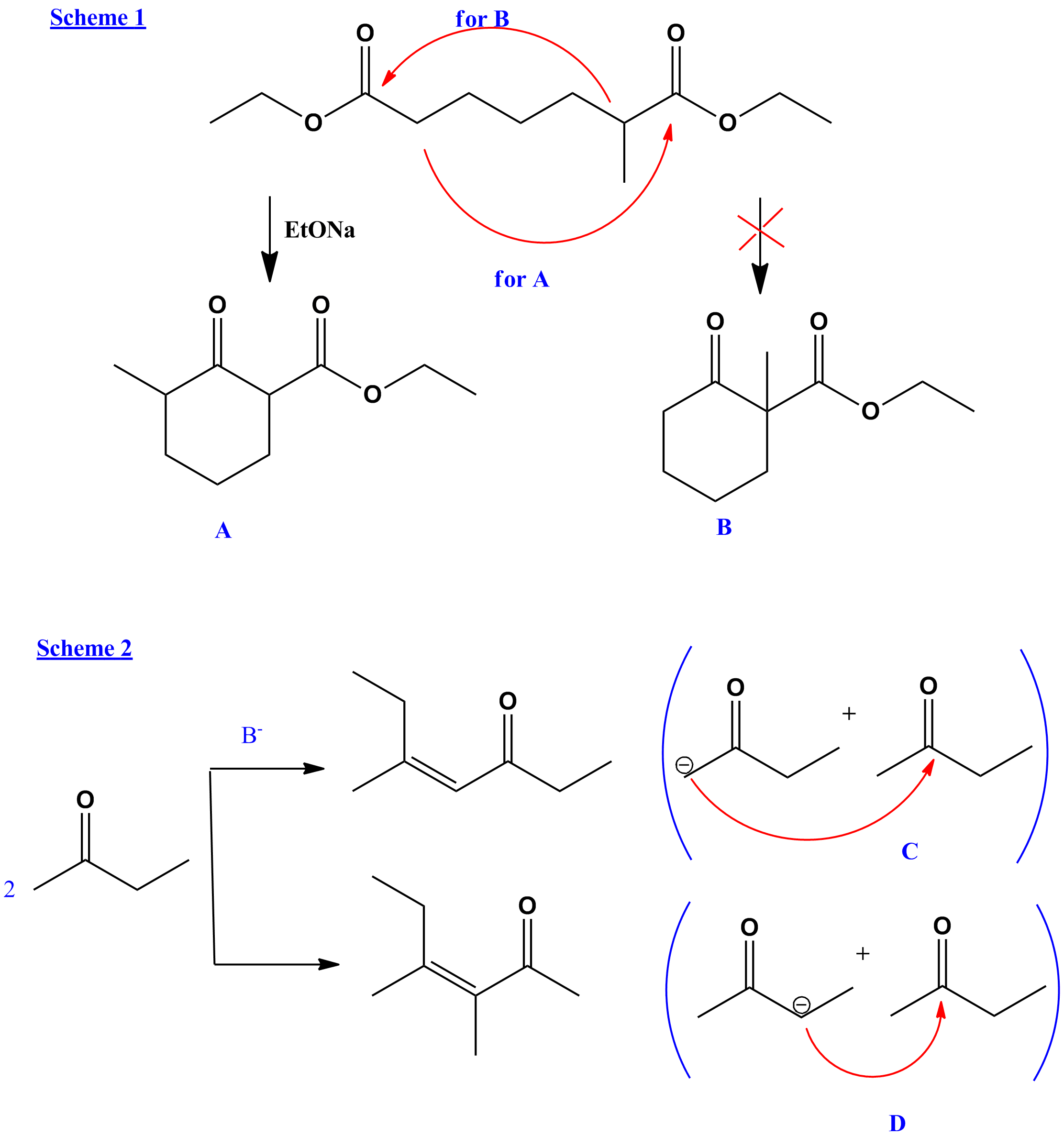
At the image above I've drawn one example for each on of them. My question is how do we choose where the deprotonation will occur. My approach was that the base will deprotonate the substrate in order to produce the most stable enolate. In the Claisen condensation, A should be the main product since the -CH2- group is present thus corresponding the mechanism. For B there will be no acidic hydrogens so it is not favoured, or at least that's what I thought.
Moving to the aldol condensation I can't figure out which product will be the main. Theoretically in C, the enolate is more stable than that of D, but the final unsaturated ketone isn't more stable in D?
Either way, I can't comprehend how can we determine what enolate will be produced each time.