What Fleming is likely referring to is work by Edward Thornton (J. Am. Chem. Soc. 1972, 94, 1168) in which he attempted to make use of secondary kinetic isotope effects (KIE) to prove whether the Diels Alder reactions being studied were indeed pericyclic, or occurred via a radical pathway.*
The phrasing in your post (which I assume is from the book itself) isn't particularly helpful. By 'geometric', Thornton was actually talking about the geometric mean and the arithmetic mean as ways of considering the isotope effects.
The reaction they actually studied was the retro Diels-Alder of 9,10-dihydro-9,10-ethanoanthrace, with varying levels of deuteration (none, di, tetra) as shown in the figure below.
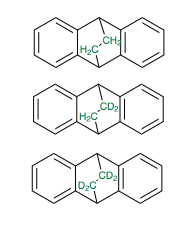
In order to discuss whats going on, we need to define some terms:
- In the reaction, two bonds are being broken, lets call those bonds A and B. Breaking bond A has a KIE of fA and breaking bond B has a KIE of fB
- Each molecule (no deuteration, di-, tetra-) has an associated rate constant, lets call these k0, k2, k4 (with the subscript indicating how many deuteriums we have).
We can now consider the calculation that Thornton went through to determine if the reaction is indeed a concerted one:#
- In a completely concerted reaction, intuitively fA = fB, since the TS is symmetrical. If the reaction is non-concerted (aka stepwise), fA cannot equal fB unless there is symmetry to the TS.
- Considering the di-deuterated molecule, two pathways are possible (breaking A or B first) to give a non-symmetrical intermediate. Thornton defines rate equations for these two as fAk0/2 and fBk0/2. We can do this since k0 is the same no matter which bond is broken in the undeuterated starting material (with the f modifying it to take into account the KIE).
- k2 (the rate constant for the retro Diels-Alder of the di-deuterated starting material) can therefore be thought of as the sum of two pathways (break A then B or B then A). Mathematically: k2 = k0(fA + fB)/2. Crucially, this relationship is valid independent of whether the reaction is concerted or stepwise.
- k4 (the rate constant for the retro Diels-Alder of the tetra-deuterated starting material) is able to be obtained similarly. Mathematically: k4 = fA fBk0.
- At this point, Thornton provides the result of solving the two previous equations simultaneously, which provides a relationship as below:
$$f_\mathrm{A},f_\mathrm{B} = \frac{k_2}{k_0} \pm \left[\left(\frac{k_2}{k_0}\right)^2 - \frac{k_4}{k_0}\right]^{1/2}$$
*: There are many examples of Diels Alder reactions that are stepwise, via radical mechanisms. The KIE's measured are necessarily small, meaning its often difficult to know if what you're looking at is actually correct. One thing that does hold up generally is that the reactions aren't ionic, due to the lack of any real solvent dependance (if the reactants are soluble in the solvent and you heat it hot enough, they generally proceed at the same rate, suggesting no stabilisation of 'intermediates' in polar solvents).
#: I'm paraphrasing the original paper, which will hopefully name it more digestible, but I still encourage you to read the actual thing, despite it being fairly dense