This is a rather intellectually-stimulating question and one that is also very difficult to answer. You have constructed a very good case for why the nucleophilicity order would not be expected to reverse in polar aprotic solvents, i.e. the order of intrinsic nucleophilicities of the halide ions should be $\ce {I^- > Br^- > Cl^- > F^-}$, based on the concepts of frontier molecular orbital interactions, as well as electronegativity and polarisability of the nucleophilic atom.
Affirming your viewpoint
Admittedly, the saturated carbon atom is a soft centre and based on electronegativity considerations, the energy of the $\ce {HOMO}$ of the halide ions should increase from $\ce {F^-}$ to $\ce {I^-}$, i.e. the hardness of the the halide ions as nucleophiles decreases from $\ce {F^-}$ to $\ce {I^-}$. Based on the hard-soft acid-base (HSAB) principle, we would expect the the strength of the interaction between the halide ion and the carbon centre to increase from $\ce {F^-}$ to $\ce {I^-}$.
We can provide more quantitative justification for this using the Klopman-Salem equation$\ce {^1}$:
$$\Delta E=\frac{Q_{\text{Nu}}Q_{\text{El}}}{\varepsilon R}+\frac{2(c_{\text{Nu}}c_{\text{El}}\beta)^2}{E_{\text{HOMO}(\text{Nu})}\pm E_{\text{LUMO}(\text{El})}}$$
The first term is indicative of the strength of the Coulombic interaction while the second term is indicative of the strength of the orbital interactions. In the reaction with the soft saturated carbon, it is actually expected for the reactivity to increase from $\ce {F^-}$ to $\ce {I^-}$ (ref 1, p. 117).
At the moment, all the evidence presented points us towards the conclusion that you have posited. However, we have overlooked the importance of a thermodynamic factor - the strength of the $\ce {C-X}$ bond.
A not-particularly-relevant add-on
Actually, if you think about it the considerations made in the Klopman-Salem equation are essentially kinetic considerations. We cannot conclude anything about reaction energetics from such considerations. The strength of the $\ce {HOMO-LUMO}$ interaction or that of the Coulombic attraction tells us nothing about how the electrons would organise themselves subsequently, what would be the strength of the resultant bonds etc.
A comment made on this by Olmstead & Brauman (1977)
On this topic of explaining the relative order of intrinsic nucleophilicity of the halide ions, Olmstead & Brauman (1977) had this to say$\ce {^2}$:
The intrinsic nucleophilicities follow the reverse order of the polarizabilities (e.g., $\ce {CH3O^-}$ > $\ce {CH3S^-}$ and $\ce {F^-}$ > $\ce {Cl^-}$ > $\ce {Br^-}$). This could be due to a stronger interaction between the more concentrated molecular orbitals of the anion with the carbon center. It could also be simply a reflection of the greater thermodynamic methyl cation affinities of the smaller anions.
It is important to note that Olmstead & Brauman are merely postulating the reasons. However, I do feel that the point on thermodynamics leads us on the right track to answering this question. On the other point of "more concentrated MOs", I am unclear of how it can be interpreted.
Let us focus now on this point.
How does thermodynamics even influence kinetics?
Well... It certainly does here. What are we concerned with when dealing with nucleophilicity? As it is a kinetic phenomenon, we are essentially concerned with the rate of the substitution reaction. This makes the consideration of the activation energy ($E_\mathrm{a}$) a particularly important one because the higher the $E_\mathrm{a}$, the lower the rate of the substitution reaction.
Here, the $E_\mathrm{a}$ can be taken to be the difference in energy between the reactants and the transition state. Thus, we can decrease the $E_\mathrm{a}$ by either raising the energy level of reactants or decreasing the energy level of the transition state. The reaction energy profile for an $\ce {S_N2}$ reaction is shown below, taken from ref. 3 (p. 394).
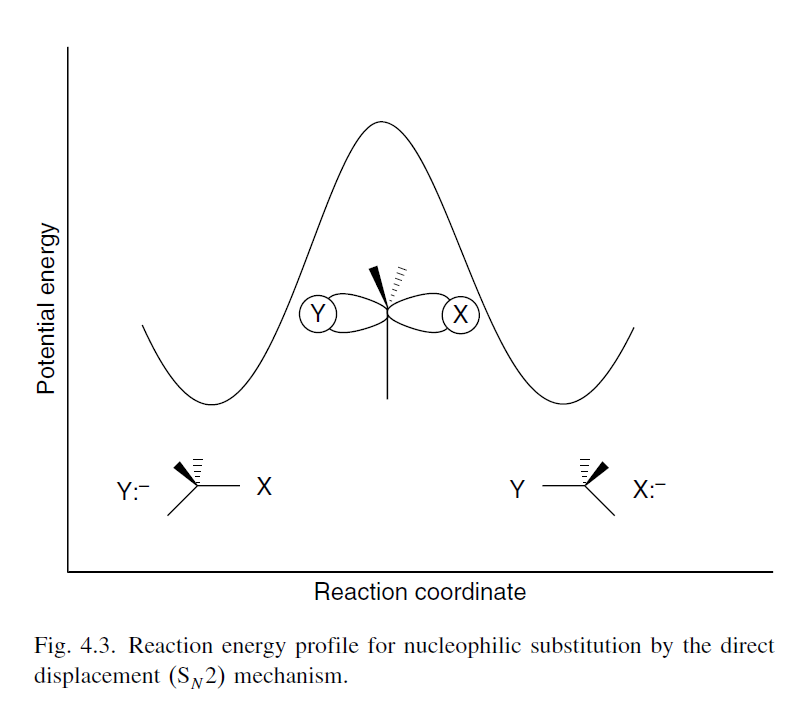
There are two ways to see how the strength of the $\ce {C-X}$ bond can affect the $E_\mathrm{a}$. I am not sure if they may be considered equivalent perspectives. Nonetheless, I will present both.
The first perspective is well-explained by Francis & Sundberg (2007) with regard to the $\ce {S_N2}$ reaction$\ce {^3}$:
Because the $\ce {S_N2}$ process is concerted, the strength of the partially formed new bond is reflected in the TS. A stronger bond between the nucleophilic atom and carbon atom results in a more stable TS and a reduced activation energy.
Another perspective (which may be somewhat similar to the first, or even possibly the same) is that the transition state structure bears resemblance to the product structure, based on the Hammond's postulate. However, this would be more applicable for an endothermic $\ce {S_N2}$ reaction where the transition state structure does indeed bear more resemblance to the product. When the product is more stable, i.e. has bonds that form more exothermically, the energy of the transition state would thus be lowered, and the $E_\mathrm{a}$ would also decrease, although not necessarily proportionally.
Conclusion
The argument you have presented failed to consider the important thermodynamics factor. Thus, it resulted in the predicted reactivity of the halide ions in the reversed order. The takeaway here is that the application of the $\ce {HSAB}$ concept alone does not always produce the correct prediction.
Additional information
More examples are cited for which the HSAB concept fails. These include:
- The favourable combination of $\ce {H^+}$, a hard acid, and $\ce {H^-}$, a soft base (discussed here as well) (ref. 4).
- The fact that the rate of reaction between $\ce {Ag^+}$, soft acid, and an alkene, soft base, is slower than that between $\ce {Ag^+}$ and $\ce {OH^-}$, a hard base (ref. 1, p. 110)
References
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions (Student Edition). John Wiley & Sons, Ltd. United Kingdom, 2009.
- Olmstead, W. N.; Brauman, J. I. Gas-phase nucleophilic displacement reactions. J. Am. Chem. Soc. 1977, 99(13), 4219–4228.
- Carey, F. A.; Sundberg, R. J. Advanced Organic Chemistry Part A. Structure and Mechanisms (5th ed.). Springer, 2007.
- Pearson, R. G.; Songstad, J. Application of the Principle of Hard and Soft Acids and Bases to Organic Chemistry. J. Am. Chem. Soc. 1967, 89(8), 1827-1836.