So let's say for a given molecule I have the volume of a unit cell, the shape of the unit cell (monoclinic, cubic, etc), the cell angles alpha, beta, and gamma, the cell lengths a,b, and c, and a list of atoms with their corresponding fractional units. How do I convert these fractional coordinates of the atoms into cartesian coordinates for any given cell shape? Orthogonal or not?
$\begingroup$
$\endgroup$
2
-
$\begingroup$ ruppweb.org/Xray/tutorial/… The mathematics behind it is explained briefly here. I wrote an excel sheet some years ago to perform that transformation, if you need it I can see if I can find it $\endgroup$– user32223Commented Jul 21, 2020 at 6:54
-
$\begingroup$ A bit late but I wrote this to perform the calculations: ic50.org/fractorth Feel free to check it, thank you! $\endgroup$– batCommented Apr 6, 2021 at 23:17
Add a comment
|
3 Answers
$\begingroup$
$\endgroup$
6
You have to use a matrix to convert. This is derived in most textbooks on crystallography, such as McKie & McKie 'Essentials of Crystallography'.
The matrix is
$$ M=\begin{bmatrix} a & 0 & \\b\cos(\gamma)& b\sin(\gamma) & 0\\c\cos(\beta) & cn_2 & c\sqrt{\sin^2(\beta)-n_2^2} \end{bmatrix}$$
where $$n_2=\frac{\cos(\alpha)-\cos(\gamma)\cos(\beta)}{\sin(\gamma)}$$
and $a,b,c $ are the unit cell dimensions and $\alpha,\beta,\gamma$ the angles in radians. The determinant of $M$ is the cell volume.
If $V_1$ is the vector of fractional cell coordinates $x/a,y/b,z/c$ etc. the matrix multiplication calculation $\hat d_{12}=(V_1-V_2)\cdot M $ and then $L_{12}=\sqrt{ \hat d_{12}\cdot \hat d_{12}}$ gives the bond length atom 1 to 2. The angle $\theta$ between two bonds 12 and 23 is given by a dot product $\cos(\theta)=\hat d_{12}\cdot \hat d_{23}/(L_{12}L_{23})$
An example calculation in python is shown below for a triclinic crystal. The @ is matrix multiplication.
import numpy as np
a = 7.55 # cell parameters
b = 4.99
c = 12.50
alpha = 122.5*np.pi/180
beta = (95+18/60)*np.pi/180
gama = (118+54/60)*np.pi/180
V1 = np.array( [-0.2812 , -0.0628 , 0.1928 ] ) # x/a, y/a, z/a
V2 = np.array( [-0.2308 , -0.0972 , 0.2931 ] )
V3 = np.array( [-0.3639 , -0.1913 , 0.3521 ] )
n2 = (np.cos(alpha)-np.cos(gama)*np.cos(beta))/np.sin(gama)
M = np.array([[a,0,0],[b*np.cos(gama),b*np.sin(gama),0],
[c*np.cos(beta),c*n2,c*np.sqrt(np.sin(beta)**2-n2**2)]])
dcm1 = (V1-V2) @ M # row x matrix
L12 = np.sqrt(dcm1 @ dcm1) # sqrt(dot product)
L12
-
$\begingroup$ Is the matrix set up to multiply from the right or from the left? I'm more familiar with vector times matrix rather than matrix times vector. $\endgroup$– Karsten ♦Commented Jul 22, 2020 at 16:28
-
$\begingroup$ It's allowed if the 1D matrix (row vector) is of length n and the matrix n x n so the columns of the vector (n) is the same length as the rows of the matrix (n). $\endgroup$ Commented Jul 22, 2020 at 17:06
-
1$\begingroup$ I've added some code so you can see how it works. $\endgroup$ Commented Jul 22, 2020 at 17:20
-
$\begingroup$ It is allowed, but it gives a different answer unless you switch to the transpose when you switch the order, math.stackexchange.com/a/1623117/657106 $\endgroup$– Karsten ♦Commented Jul 23, 2020 at 0:31
-
$\begingroup$ yes, this is what I meant by using a row vector. $\endgroup$ Commented Jul 23, 2020 at 6:21
$\begingroup$
$\endgroup$
3
The answer by porphyrin is correct and arguably the proper way to perform the transformation. But there is another / simpler way to convert between fractional and cartesian coordinates that requires a little less programming.
Provided you have the lattice vectors of the cell $\vec{a}_1$, $\vec{a}_2$, and $\vec{a}_3$ (in cartesian coordinates), you can organize these into a matrix with the lattice vectors stored column-wise:
$$A = \begin{bmatrix}\vec{a}_1 & \vec{a}_2 & \vec{a}_3\end{bmatrix} = \begin{bmatrix} a_{11} & a_{21} & a_{31} \\ a_{12} & a_{22} & a_{32} \\ a_{13} & a_{23} & a_{33} \end{bmatrix}$$
If we denote an atomic position in cartesian space as $\vec{x}$ and the corresponding point in fractional (or crystal) coordinates as $\vec{y}$, we can convert between the two using the following equation:
$$\vec{x} = A\vec{y}$$
To convert from cartesian coordinates to fractional coordinates, you can just trivially invert this equation to get:
$$\vec{y} = A^{-1}\vec{x}$$
So, now if you have a set of $m$ atomic positions in fractional coordinates, you can organize them into an $m\times 3$ matrix denoted as $Y$. To get the corresponding set of atomic positions in cartesian coordinates denoted as $X$ (also an $m\times 3$ matrix), you can use the following equation:
$$X = (AY^T)^T$$
and similarly to convert a set of cartesian coordinates to fractional coordinates you can use the inverse relation:
$$Y = (A^{-1}X^T)^T$$
In Python you can do the following:
import numpy as np
# Pick your favorite lattice vectors
a1 = np.array([...])
a2 = np.array([...])
a3 = np.array([...])
# Build the matrix of lattice vectors stored column-wise
# and get its inverse
A = np.vstack([a1, a2, a3]).T
A_inv = np.linalg.inv(A)
# A random set of fractional coordinates stored row-wise
Y = np.array([[0.25, 0.75, 0.1],
[0.75, 0.25, 0.3],
[0.25, 0.75, 0.5]]
)
# Compute the cartesian coordinates
X = np.matmul(A, Y.T).T
# Perform the inverse operation to get fractional coordinates
Y_ = np.matmul(A_inv, X.T).T
You should be able to confirm that the arrays Y and Y_ are the same.
-
$\begingroup$ The lattice vectors should be given in cartesian coordinates, otherwise it does not work. The inverse of matrix A is the matrix M in the answer by @porphyrin. $\endgroup$– Karsten ♦Commented Jul 29, 2021 at 16:02
-
2$\begingroup$ Yes, the lattice vectors must be in cartesian coordinates. And yes, the inverse of matrix A is the metric matrix M. In my opinion this approach is a bit easier to remember, but it is entirely equivalent to the method described by porphyrin. $\endgroup$– StephenCommented Jul 29, 2021 at 16:43
-
$\begingroup$ Seconded - life just becomes so much easier if you just work with the lattice vectors rather than mucking about with angles. $\endgroup$– Ian BushCommented Jul 29, 2021 at 18:37
$\begingroup$
 $\endgroup$
$\endgroup$
Without doubt, the previously provided answers will work well if the crystallographic model is a description in space group triclinic $P1$. However on occasion, the symmetry of the unit cell is higher and the molecule of interest is on a special position, for example a centre of inversion. As a result, the underlying .cif file need not to contain as many explicit coordinate tuples as atoms in the molecule. On the other hand, on first glance by visual inspection, the model might appear incomplete. This answer aims to complement the previous answers from the perspective not wanting to write new programs.
Entry COD 2102215 of the COD about 2,4,6-trimethoxy-1,3,5-triazine serves as an example. The initial display (of the asymmetric unit) by the COD and the complete molecular structure are provided below:
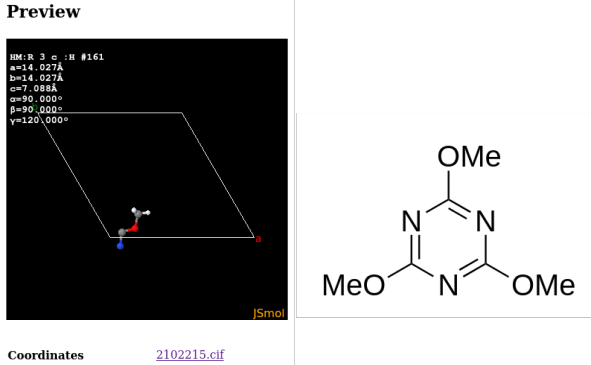
The .cif describes only coordinate tuples for seven atom as such, which is exactly a third of the molecule. This is because the threefold symmetry axis of the molecule aligns with a threefold axis of the unit cell. A script like codcif2sdf, part of the cod-tools collection, however is able to recognize the described symmetries of the unit cell acting on the molecule, and to join the molecular fragments into a .sdf:
codcif2sdf 2102215.cif > 2102215.sdf
The later format is one many other programs accept as input. Equally, its content may be rewritten in other formats like the most elementary structure format .xyz, or to yield visualizations e.g., by OpenBabel
obabel -isdf 2102215.sdf -oxyz -O 2102215.xyz
obabel -isdf 2102215.sdf -opng -O 2102215.png
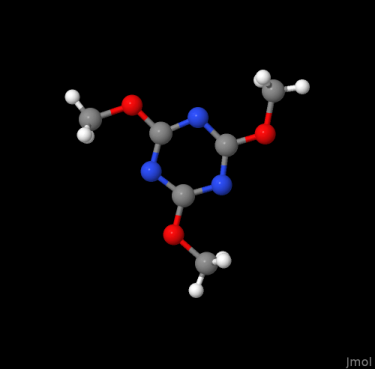
Further details about OpenBabel are available here.
answered Jul 29, 2021 at 18:01