With your reply you seem to have answered part of your question. I would like to add a bit about the other part. I will have to do it mathematically first, there is no other shortcut, but then we will come back and try to connect the idea. I will be precise on where the thermodynamic properties are evaluated, using parenthesis everywhere.
[OP] In other words, how are (1) and (2) related to each other?
The connection between Eqs. (1) and (2) was established by Gibbs himself, it is named Gibbs theorem. Simply put, he was trying to connect the properties of the pure species (as ideal gases) to the properties of the mixture (still an ideal gas). One possible formulation is the following:
A partial molar property of a species in an ideal-gas mixture is equal to its molar property as a pure substance, evaluated at the temperature of the mixture but at its partial presure in the mixture. The exception of this is the partial molar volume.
In mathematical form, for a generic thermodynamic property of species $j$ named $M_j$, we have
$$ \bar{M}_j^\pu{ig}(p,T) = M_j^\pu{ig}(p_j,T) \tag{1} $$
Eq. (1) is almost all that we need to obtain the Eq. (2) of your post. The other one is the summability relation that connects a thermodynamic property $M$ (of the whole mixture) to the partial molar property of all the species in the mixture
$$ M(p,T,y_1, y_2,..., y_N) = \sum_k y_k \bar{M}_k(p,T,y_1, y_2,..., y_N) \tag{2} $$
Eq. (2) is general and is not restricted by the behaviour imposed over the mixture. The derivation of this equation is not that important here.
We will calculate the partial molar enthalpy, partial molar entropy, and then obtain the partial molar Gibbs energy by its definition.
- The enthalpy is easy, because for an ideal gas the enthalpy is independent of the pressure, and by Gibbs theorem we have
$$ \bar{H}_j^\pu{ig}(p,T) = H_j^\pu{ig}(p_j,T) = H_j^\pu{ig}(p,T) \tag{3} $$
- Applying Gibbs theorem to the entropy yields
$$ \bar{S}_j^\pu{ig}(p,T) = S_j^\pu{ig}(p_j,T) \tag{4} $$
We would like to have evaluated the right-hand side at $p$ rather than $p_j$. This can be done by remembering the entropy of an ideal gas as a function of pressure and temperature, and evaluating the change for an isothermal process at temperature $T$
\begin{align}
\require{cancel}
dS_j^\pu{ig} &= \frac{c_\pu{P}^\pu{ig}}{T} \; dT - \frac{R}{p} \; dp \\
dS_j^\pu{ig} &= -\frac{R}{p} \; dp \\
\int_{S_j^\pu{ig}(p_j,T)}^{S_j^\pu{ig}(p,T)} dS_j^\pu{ig} &=
-R\int_{p_j}^{p} \frac{dp}{p} \\
S_j^\pu{ig}(p,T) - S_j^\pu{ig}(p_j,T) &= -R\ln\left(\frac{p}{p_j}\right) \\
S_j^\pu{ig}(p_j,T) &= S_j^\pu{ig}(p,T) + R\ln\left(\frac{p}{p_j}\right) \\
S_j^\pu{ig}(p_j,T) &= S_j^\pu{ig}(p,T) + R\ln\left(\frac{\cancel{p}}{y_j \cancel{p}}\right) \\
S_j^\pu{ig}(p_j,T) &= S_j^\pu{ig}(p,T) - R\ln(y_j) \tag{5}
\end{align}
In the last line I just related the partial pressure to the pressure of the mixture. Combining Eqs. (4) and (5)
$$ \bar{S}_j^\pu{ig}(p,T) = S_j^\pu{ig}(p,T) - R\ln(y_j) \tag{6} $$
We can obtain the partial molar Gibbs energy just by definition, multiplying both sides by the total number of moles $n$, and then differentiating both sides at constant $p$, $T$, and all moles except the species $j$
\begin{align}
G &= H - TS \\
nG &= nH - TnS \\
\left[\frac{\partial (nG)}{\partial n_j}\right]_{p,T,n_k\neq j} &=
\left[\frac{\partial (nH - TnS)}{\partial n_j}\right]_{p,T,n_k\neq j} \\
\bar{G}_j &= \left[\frac{\partial (nH)}{\partial n_j}\right]_{p,T,n_k\neq j}
-T\left[\frac{\partial (nS)}{\partial n_j}\right]_{p,T,n_k\neq j} \\
\bar{G}_j &= \bar{H}_j - T\bar{S}_j \tag{7}
\end{align}
This equation is general, not resctricted to an ideal-gas behaviour. Now we can combine Eqs. (3), (6), and (7) to obtain the partial molar Gibbs energy (note that between lines one and two we use Gibbs theorem over the enthalpy and entropy)
\begin{align}
\bar{G}_j^\pu{ig}(p,T) &= \bar{H}_j^\pu{ig}(p,T) - T\bar{S}_j^\pu{ig}(p,T) \\
\bar{G}_j^\pu{ig}(p,T) &= H_j^\pu{ig}(p_j,T) - TS_j^\pu{ig}(p_j,T) \\
\bar{G}_j^\pu{ig}(p,T) &= H_j^\pu{ig}(p,T) -
T[S_j^\pu{ig}(p,T) - R\ln(y_j)] \\
\bar{G}_j^\pu{ig}(p,T) &= [H_j^\pu{ig}(p,T) -
TS_j^\pu{ig}(p,T)] + RT\ln(y_j) \\
\bar{G}_j^\pu{ig}(p,T) &= G_j^\pu{ig}(p,T) + RT\ln(y_j) \tag{8}\\
\end{align}
Finally we combine the summability relation Eq.(2) with Eq. (8), since we are summing, now we convert the subscript $j$ to $k$
\begin{align}
G^\pu{ig}(p,T, y_1, y_2, ..., y_N) &= \sum_k y_k [G_k^\pu{ig}(p,T) + RT\ln(y_k)] \\
G^\pu{ig}(p,T,y_1, y_2, ..., y_N) &= \sum_k y_k G_k^\pu{ig}(p,T) + RT\sum_k y_k\ln(y_k) \\
G^\pu{ig}(p,T, y_1, y_2, ..., y_N) - \sum_k y_k G_k^\pu{ig}(p,T) &= RT\sum_k y_k \ln(y_k)
\end{align}
the left side, by definition is the Gibbs free energy of mixing. We are subtracting the value of the Gibbs energy of the mixture and the mole-fraction-weighted Gibbs energy of all the species involved. Finally
$$ \boxed{\Delta_\pu{mix}G = RT\sum_k y_k \ln(y_k) <0} \tag{9} $$
[OP] So if the two gases are independent, why is there a Gibbs energy of mixing? Shouldn't there be a requirement for an interaction energy in order to see a difference in Gibbs energy before and after mixing?
Even in the absence of intermolecular forces between the species, there is a change in the Gibbs energy when we mix them. IMHO (I cannot prove this), this was what Gibbs foresaw before doing all this:
- He knew that for an ideal gas the enthalpy will not be modified. Thus, for a mixing at constant $p$ and $T$, there will not be heat exchanged with the surroundings.
- He knew that the mixing could not violate the second law.
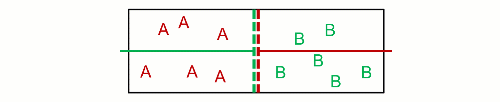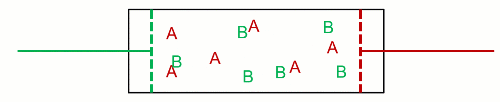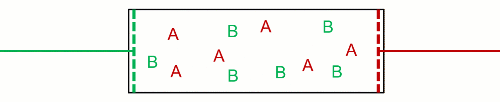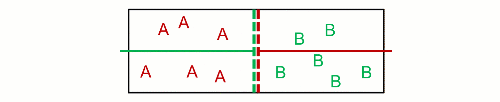
Therefore, he visioned $N$ non-interacting particles separated at $p$ and $T$, and then they got mixed at $p_k$ and $T$. This decrease in pressure is the effect wanted by Gibbs, because it inevitably increases the entropy. As a consequence, it inevitably decreases the Gibbs free energy (his energy), and mixing of ideal gases is always a spontaneous process. Likewise, unmixing is also always a non-spontaneous process. This is really near to what Poutnik said in the comments when he made a difference between both pressures:
[Poutnik] When you joint the same containers with 2 gases, at the same T and p, than partial p of both gases decreases to 1/2 of their initial p, therefore their chem. potential decreases as well.
[OP] How could you turn the process of mixing into a reversible process and show the maximal work that is done?
You visualized this in the images in your post. The calculation of the maximum work is automatic. The maximum work that can be done by the system to the surroundings at the same temperature $T$ as the system is
$$ \boxed{W^\pu{ideal} = \Delta_\pu{mix} G^\pu{ig} < 0} \tag{11} $$
so mixing of ideal gases can be used to obtain work from the outside.
Final Comment When I said that there was a connection between Eqs. (1) and (2), it is implicit in here, but we can show it more clearly. For a constant temperature process and a pure substance we have
\begin{align}
dG_j^\pu{ig} &= V_j^\pu{ig}dp - S_j^\pu{ig}dT \\
dG_j^\pu{ig} &= V_j^\pu{ig}dp \\
dG_j^\pu{ig} &= \frac{RT}{p} dp \\
\int dG_j^\pu{ig} &= RT
\int \frac{dp}{p} \\
G_j^\pu{ig}(p,T) &= G_{j,0}^\pu{ig} + RT\ln(p) \\
\end{align}
The jump to your Eq. (1), is by Gibbs theorem in order to change $p$ for $y_jp$
\begin{align}
\bar{G}_j^\pu{ig}(p,T) &= \mu_{j,0}^\pu{ig} + RT\ln(y_jp) \\
\mu_j^\pu{ig}(p,T) &= \mu_{j,0}^\pu{ig} + RT\ln(y_jp) \\
\end{align}
If not, there is no way we can write your equation (with $p_\mathrm{A}$ in your case), and arrive at an expression for the partial molar Gibbs energy, also known as the chemical potential.