Problem description
I have a substance that is extremely non-polar and therefore does not ionize in the ESI source, which makes it impossible to quantify small amounts. The substance contains only two functional groups, which are a point of attack for possible derivatization. One is an alkyne, and the other is a hydroxyl group. Both groups are attached to the same carbon. Since the substance is to be detected from blood plasma, I decided against derivatization at the hydroxyl group, since numerous side reactions with proteins, which also carry a lot of hydroxyl groups, are to be expected. The alkyne, however, is a good point of attack and is suitable for bioorthogonal reactions. A professor once said about the copper(I) catalyzed alkyne-azide cycloaddition (CuAAC) that this is a reaction that "any fool could manage". I have been trying to get the reaction going for a few days now, but so far, I have not been able to detect any product in LC-MS... Decide for yourself what this says about me as a chemist.
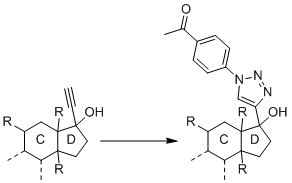
Since I have followed several approaches from papers, I slowly ask myself if my substance is not suitable for a CuAAC. Since the research project and the substance are new and unpublished, I cannot provide a complete structural formula here. But this much can be revealed: It is essentially a steroid, which carries a hydroxy group and an alkyne on carbon C17.
What was done
I performed the reaction with the pure substance (i.e. without blood plasma) according to Presolski et al., but I did not use THPTA nor aminoguanidine. All concentrations were used as described in the paper and the reaction was performed at 40°C according to the suggestion under "Troubleshooting". The azide was dissolved in DMF. Thus, there were 10 µL DMF (5%) in the buffer solution.
I also performed the reaction with the pure substance according to Hong et al.. Again, the proposed concentrations were used but neither THPA nor aminoguanidine were used.
I also tested different variations of those protocols with much longer reaction times (8 to 24 hours), more or less temperature, with and without pH 7 buffer.
As far as I know, the 4-substituted 1,2,3-triazoles formed by CuAAC should be extremely stable, even to metabolic degradation (Agalave et al.), so I do not believe that they decomposed so quickly that they were all destroyed by the measurement. To avoid ion suppression, I purified the reaction after the one-hour reaction time by RP-SPE, using the SigmaAldrich/Merck protocol.
Questions
- Does anyone have experience with CuAAC reactions that do not want to run?
- Which parameters should I at best adjust to make the reaction successful?
- Is it possible that a hydroxide in alpha-position disturbs the reaction? I haven't found anything on this yet.
- What advice is there to make the reaction successful even at very low (< 10 pg/mL) substance concentrations?
Update as of 24/07/2020
The reaction was, in fact, successful when applying the already cited protocol by Presolski et al. and using a DMF-water-ratio of 3:1 and incubating at 60 °C for 2 h. Unfortunately, though, the intensities are pretty weak which means that the reaction turnover is not great. The peak of my original substance is at 9.6 × 107 (50 µM concentration) and the intensity of the derivatized product, which should be way better ionized, is 4.2 × 107.
New question
- How to get a better turnover while maintaining a reasonably short reaction time?