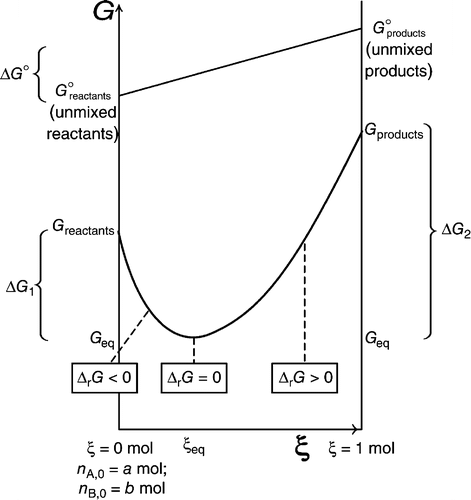
It is well-known that a reaction is spontaneous if $\Delta G < 0$. This requirement is derived from thermodynamic considerations, as done here. However, there seems to be two different ways of calculating $\Delta G$. As provided by Orthocresol in the answer here, $\Delta G$ is the rate of change of the Gibbs free energy of the reaction mixture with respect to the extent of reaction. This definition based on the rate of change cannot be disputed as a negative gradient of the graph would mean that as the forward reaction proceeds, Gibbs free energy is being minimised.
However, there appears to be another way in which $\Delta G$ is being calculated. In this approach, we take $\Delta G$ as the absolute change in Gibbs free energy going from reactants to products. One way of calculating this would be using $\Delta G = \Delta G_f [Products] - \Delta G_f [Reactants]$. Another would be $\Delta_rG = \Delta_rH - T \Delta_rS$. In this case, we are not calculating the gradient of the $G$ against extent of reaction curve but instead, we are simply calculating the absolute change in $G$ from all reactants to all products.
Clearly, the two approaches, one based on the gradient of the curve and one based on the absolute change of $G$ between reactants and products, are not the same. However, both approaches can be used to calculate $\Delta G$ in the determination of reaction spontaneity. The second approach clearly being more dubious than the first.
My attempt is rationalise this is as follows:
Both approaches are based on the idea of minimisation of the Gibbs free energy. However, they differ in their analysis of the reaction. The first approach of calculating $\Delta G$ considers an ensemble of molecules going from reactants to products. It looks at small adjustments to the ratio of reactants to products in the reaction mixture as reaction progresses and subsequent changes to entropy of the mixture (due to mixing of reactants and products)
However, in the second approach, we are trying to look at the energetics in the one-step change from a reactant molecule to a product molecule and we are merely concerned with the question: does $G$ decrease when a reactant molecule changes into a product molecule? Clearly, this process does not occur in small steps but occurs at once, unlike the conversion of a mole of reactants to a mole of products. We make use of the $\Delta G_f$ values for the reactants and products to make an approximation for the conversion of one reactant to one product. This 2nd approach is certainly not exact but it is certainly still chemically correct.