There are proteins with intrinsic symmetries. For example:
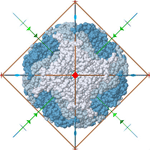
I was wondering how to use transformations such as: rotations, translation, replications to construct possible structures using 2 types of proteins.
Assume I found a binding site of the 2 proteins using Molecular Dynamics simulators.
For example the above one and this one:
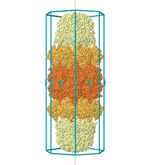
- Can I use group theory way to find possible configurations of the 2 proteins?
- How can I use the tensor product of the symmetry groups ($G_1 \times G_2$) to find possible structures?
- What graphic/framework would you recommend do perform all the manipulations on the proteins?
- Is it possible to use the transfer to create some algebraic structure that will help to construct a mechanical configurations comprised of proteins?
- Is there any tool from Baker's Lab I can use for this task?
- Amy recommendation for a tool I could use to replicate, rotate and shift structure would be appreciated us well.
TL;DR:
My goal is to use group theory or Monte Carlo simulation driven by group theory insight to create macro molecular structures using multiple (dozens) instances of the two proteins (the LEGO building blocks). Any idea that relates symmetry, transformations, group theory to create structures is much welcome.
Clarification:
Given a binding point between 2 proteins, which software can I use to create the dimers into macro-structures like in LEGO namely only by replicating, translating and rotating the Lego pieces?