These pi-type MOs are most commonly derived using Hückel MO theory. I've never been a fan of Wikipedia's technical articles, so for further reading I'd suggest using the QMUL resources online, which are very comprehensive. $\require{begingroup}\begingroup \newcommand{ket}[1]{\left|#1\right>} \newcommand{bra}[1]{\left<#1\right|} \newcommand{braket}[1]{\left< #1 \right>}$
Theory
To begin with, MOs are obtained by solving the secular equations:
$$\mathbf{H}\mathbf{c} = E\mathbf{S}\mathbf{c}$$
The derivation of this will not be covered here (it can be found in e.g. Atkins' Molecular Quantum Mechanics), but it is worth mentioning what these things are. If we consider a polyene with $n$ atoms (e.g. for ethene, $n = 2$), then
$\mathbf{H}$ is the Hamiltonian matrix, an $n \times n$ matrix whose elements $H_{ij}$ refer to the the $i$-th and $j$-th p-orbitals: $H_{ij} = \braket{\mathrm{p}_i|H|\mathrm{p}_j}$.
$\mathbf{c}$ is a $1 \times n$ column vector of coefficients: the element $c_i$ corresponds to the coefficient of the $i$-th p-orbital. That is to say, the MO $|\psi\rangle$ corresponding to the vector $\mathbf{c}$ is given by
$$|\psi\rangle = c_1 |\mathrm{p}_1\rangle + c_2 |\mathrm{p}_1\rangle + \cdots + c_n |\mathrm{p}_n\rangle.$$
By solving the secular equations, we obtain a series of permitted vectors $\mathbf{c}$: each of these solutions correspond to one MO. In general, if there are $n$ p-orbitals used as inputs, then we will obtain $n$ permitted MOs.
Each solution $\mathbf{c}$ is associated with a particular energy $E$, which is a scalar.
$\mathbf{S}$ is the overlap matrix, whose elements are given by $S_{ij} = \braket{\mathrm{p}_i|\mathrm{p}_j}$.
Hereafter, we will simplify the notation and use $|i\rangle$ in place of $|\mathrm{p}_i\rangle$ to denote the p-orbitals.
Now, simple Hückel theory makes some key assumptions about the forms of the MOs as well as some of the integrals involved in a typical quantum chemical calculation:
- π-Type MOs are linear combinations of p-orbitals, and no other orbitals contribute (so-called "sigma-pi separation")
- The value of $\braket{a|b}$ (where $\ket{a},\ket{b}$ are p-type AOs on atoms $a$ and $b$) is $1$ if $a = b$ and $0$ otherwise
- The value of $\braket{a|H|b}$ is
$$H_{ab} = \braket{a|H|b} = \begin{cases}\alpha \text{ if } a = b \\ \beta \text{ if atom }a\text{ is bonded to atom } b \\ 0 \text{ otherwise.}\end{cases}$$
Here, $\alpha$ and $\beta$ are just some constants. We don't have exact values for them (yet), but we can say that they are both negative. With these simplifications the secular equations can be readily solved. In fact, in simple Hückel theory the matrix $\mathbf{S}$ is simply equal to the identity matrix (see rule 2 above), so it can be completely ignored in the secular equations:
$$\mathbf{Hc} = E\mathbf{c}.$$
You may recognise this an eigenvalue equation for $\mathbf{H}$ (the original form, where $\mathbf{S}$ is not necessarily equal to the identity matrix, is called a generalised eigenvalue equation).
As mentioned previously, for $n$ atoms there will be $n$ vectors $\mathbf{c}$ that will satisfy the secular equations, and $n$ corresponding values of the energy $E$. Thus, for the allyl cation (for example), there will be three column vectors $\mathbf{c}^{(1)}, \mathbf{c}^{(2)}, \mathbf{c}^{(3)}$ and three associated energies $E^{(1)}, E^{(2)}, E^{(3)}$.
Note here that $\mathbf{c}^{(1)}$ refers to the first possible solution for $\mathbf{c}$, whereas $c_1$ refers to the first component of $\mathbf{c}$ (i.e. the coefficient of p-orbital number one).
The allyl cation
Using the atom numbering scheme as shown above, and the assumptions mentioned in the previous section, we find that the Hamiltonian matrix $\mathbf{H}$ is:
$$\mathbf{H} =
\begin{pmatrix}
\alpha & \beta & 0 \\
\beta & \alpha & \beta \\
0 & \beta & \alpha
\end{pmatrix}.$$
For example, the entry $\mathbf{H}_{12} = \beta$ because atoms 1 and 2 are bonded to each other, and the entry $\mathbf{H}_{13} = 0$ because atoms 1 and 3 are not bonded to each other.
The secular equations then take the following form:
$$\begin{pmatrix}
\alpha & \beta & 0 \\
\beta & \alpha & \beta \\
0 & \beta & \alpha
\end{pmatrix}
\begin{pmatrix}
c_1 \\
c_2 \\
c_3
\end{pmatrix} = E\begin{pmatrix}
c_1 \\
c_2 \\
c_3
\end{pmatrix},$$
and the challenge is then to find the permissible vectors $\mathbf{c} = (c_1, c_2, c_3)$ which obey this equation, which are called eigenvectors. This process is outlined in many maths books, and is thus not covered here in full detail. Briefly, however, the idea is that we move the RHS over:
$$\begin{pmatrix}
\alpha - E & \beta & 0 \\
\beta & \alpha - E & \beta \\
0 & \beta & \alpha - E
\end{pmatrix}
\begin{pmatrix}
c_1 \\
c_2 \\
c_3
\end{pmatrix} = \mathbf{0},
$$
and there can only be nontrivial solutions to this problem (i.e. solutions where $\mathbf{c} \neq \mathbf{0}$) if the secular determinant (i.e. the determinant of the matrix on the LHS) is zero:
$$\begin{vmatrix}
\alpha - E & \beta & 0 \\
\beta & \alpha - E & \beta \\
0 & \beta & \alpha - E
\end{vmatrix} = 0$$
Expanding this out allows you to obtain the eigenvalues. In increasing energy (recall that $\beta < 0$), they are
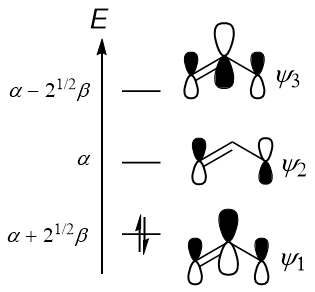
$$\begin{align}E^{(1)} &= \alpha + \sqrt{2}\beta & E^{(2)} &= \alpha & E^{(3)} &= \alpha - \sqrt{2}\beta \end{align},$$
and the corresponding eigenvectors are
$$\begin{align}
\mathbf{c}^{(1)} &= \begin{pmatrix} 1/2 \\ 1/\sqrt{2} \\ 1/2 \end{pmatrix}
&
\mathbf{c}^{(2)} &= \begin{pmatrix} 1/\sqrt{2} \\ 0 \\ -1/\sqrt{2} \end{pmatrix}
&
\mathbf{c}^{(3)} &= \begin{pmatrix} 1/2 \\ -1/\sqrt{2} \\ 1/2 \end{pmatrix}.
\end{align}$$
In other words, the lowest-energy pi-type MO, $\mathbf{c}^{(1)}$, is expressed as
$$\ket{\psi^{(1)}} = \frac{1}{2}\ket{1} + \frac{1}{\sqrt{2}}\ket{2} + \frac{1}{2}\ket{3},$$
where $\ket{i}$ is the 2p atomic orbital on carbon $i$. Note that the coefficients all have the same sign, indicating that the p orbitals are all in phase with each other. However, the coefficient for carbon 2 (the middle carbon) is larger than the others. Hence, a sketch of this MO should technically show that the middle p-orbital makes a larger contribution than the outer two p-orbitals. This is what TAR86 meant by their comment on your question:
Hückel MO theory will also give you different absolute values for the linear combination coefficients, whereas your drawing suggests the same coefficients throughout.
Anyway, likewise for the LUMO we have
$$\ket{\psi^{(2)}} = \frac{1}{\sqrt{2}}\ket{1} - \frac{1}{\sqrt{2}}\ket{3}$$
Note that: (1) the p-orbitals have different phases (different sign of coefficient), and (2) the p-orbital on carbon 2 does not contribute to the MO. This leads to the node observed in the second MO. This is why the allyl cation is electrophilic on C-1 and C-3, but not on C-2: the LUMO is $\ket{\psi^{(2)}}$, which has zero coefficient on C-2.
All in all, the MO diagram for the allyl cation is as follows:
Results for other / larger polyenes may be obtained in a similar fashion, albeit perhaps with more complications in finding the eigenvalues and eigenvectors. It is recommended to use software which can perform symbolic maths: for example, if you type eigenvalues of [[a, b, 0], [b, a, b], [0, b, a]] into WolframAlpha, then it tells you the eigenvalues (i.e. the energies)

and the eigenvectors (i.e. the MOs), which is the same as what we found above, except that the solution above is normalised (i.e. every eigenvector is divided through by a constant such that $c_1^2 + c_2^2 + \cdots + c_n^2 = 1$):

