Tetrahedral complexes
Let's consider, for example, a tetrahedral $\ce{Ni(II)}$ complex ($\mathrm{d^8}$), like $\ce{[NiCl4]^2-}$. According to hybridisation theory, the central nickel ion has $\mathrm{sp^3}$ hybridisation, the four $\mathrm{sp^3}$-type orbitals are filled by electrons from the chloride ligands, and the $\mathrm{3d}$ orbitals are not involved in bonding.
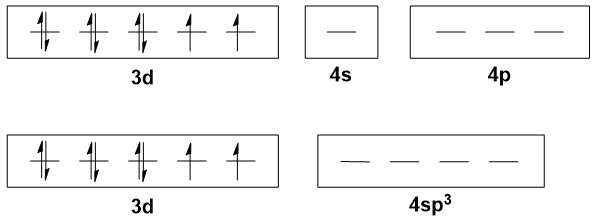
Already there are several problems with this interpretation. The most obvious is that the $\mathrm{3d}$ orbitals are very much involved in (covalent) bonding: a cursory glance at a MO diagram will show that this is the case. If they were not involved in bonding at all, they should remain degenerate, which is obviously untrue; and even if you bring in crystal field theory (CFT) to say that there is an ionic interaction, it is still not sufficient.
If accuracy is desired, the complex can only really be described by a full MO diagram. One might ask why we should believe the MO diagram over the hybridisation picture. The answer is that there is a wealth of experimental evidence, especially electronic spectroscopy ($\mathrm{d-d^*}$ transitions being the most obvious example), and magnetic properties, that is in accordance with the MO picture and not the hybridisation one. It is simply impossible to explain many of these phenomena using this $\mathrm{sp^3}$ model.
Lastly, hybridisation alone cannot explain whether a complex should be tetrahedral ($\ce{[NiCl4]^2-}$) or square planar ($\ce{[Ni(CN)4]^2-}$, or $\ce{[PtCl4]^2-}$). Generally the effect of the ligand, for example, is explained using the spectrochemical series. However, hybridisation cannot account for the position of ligands in the spectrochemical series! To do so you would need to bring in MO theory.
Octahedral complexes
Moving on to $\ce{Ni(II)}$ octahedral complexes, like $\ce{[Ni(H2O)6]^2+}$, the typical explanation is that there is $\mathrm{sp^3d^2}$ hybridisation. But all the $\mathrm{3d}$ orbitals are already populated, so where do the two $\mathrm{d}$ orbitals come from? The $\mathrm{4d}$ set, I suppose.
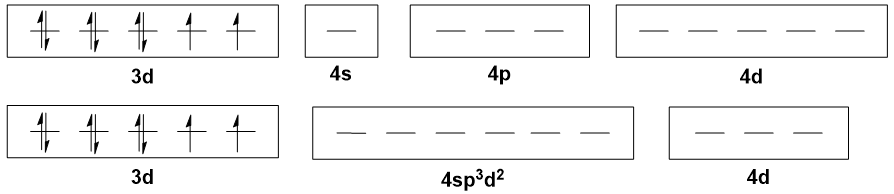
The points raised above for tetrahedral case above still apply here. However, here we have something even more criminal: the involvement of $\mathrm{4d}$ orbitals in bonding. This is simply not plausible, as these orbitals are energetically inaccessible. On top of that, it is unrealistic to expect that electrons will be donated into the $\mathrm{4d}$ orbitals when there are vacant holes in the $\mathrm{3d}$ orbitals.
For octahedral complexes where there is the possibility for high- and low-spin forms (e.g., $\mathrm{d^5}$ $\ce{Fe^3+}$ complexes), hybridisation theory becomes even more misleading:
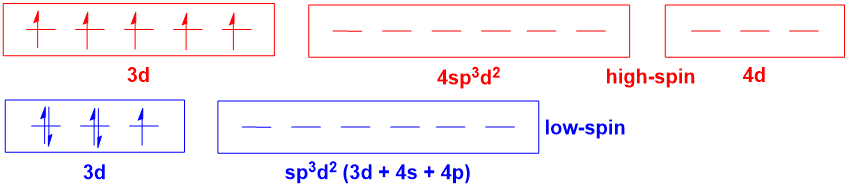
Hybridisation theory implies that there is a fundamental difference in the orbitals involved in metal-ligand bonding for the high- and low-spin complexes. However, this is simply not true (again, an MO diagram will illustrate this point). And the notion of $\mathrm{4d}$ orbitals being involved in bonding is no more realistic than it was in the last case, which is to say, utterly unrealistic. In this situation, one also has the added issue that hybridisation theory provides no way of predicting whether a complex is high- or low-spin, as this again depends on the spectrochemical series.
Summary
Hybridisation theory, when applied to transition metals, is both incorrect and inadequate.
It is incorrect in the sense that it uses completely implausible ideas ($\mathrm{3d}$ metals using $\mathrm{4d}$ orbitals in bonding) as a basis for describing the metal complexes. That alone should cast doubt on the entire idea of using hybridisation for the $\mathrm{3d}$ transition metals.
However, it is also inadequate in that it does not explain the rich chemistry of the transition metals and their complexes, be it their geometries, spectra, reactivities, or magnetic properties. This prevents it from being useful even as a predictive model.
What about other chemical species?
You mentioned that hybridisation works well for "other compounds." That is really not always the case, though. For simple compounds like water, etc. there are already issues associated with the standard VSEPR/hybridisation theory. Superficially, the $\mathrm{sp^3}$ hybridisation of oxygen is consistent with the observed bent structure, but that's just about all that can be explained. The photoelectron spectrum of water shows very clearly that the two lone pairs on oxygen are inequivalent, and the MO diagram of water backs this up. Apart from that, hybridisation has absolutely no way of explaining the structures of boranes; Wade's rules do a much better job with the delocalised bonding.
And these are just Period 2 elements - when you go into the chemistry of the heavier elements, hybridisation generally becomes less and less useful a concept. For example, hypervalency is a huge problem: $\ce{SF6}$ is claimed to be $\mathrm{sp^3d^2}$ hybridised, but in fact $\mathrm{d}$-orbital involvement in bonding is negligible. On the other hand, non-hypervalent compounds, such as $\ce{H2S}$, are probably best described as unhybridised - what happened to the theory that worked so well for $\ce{H2O}$? It just isn't applicable here, for reasons beyond the scope of this post.
There is probably one scenario in which it is really useful, and that is when describing organic compounds. The reason for this is because tetravalent carbon tends to conform to the simple categories of $\mathrm{sp}^n$ $(n \in \{1, 2, 3\})$; we don't have the same teething issues with $\mathrm{d}$-orbitals that have been discussed above. But there are caveats. For example, it is important to recognise that it is not atoms that are hybridised, but rather orbitals: for example, each carbon in cyclopropane uses $\mathrm{sp^5}$ orbitals for the $\ce{C-C}$ bonds and $\mathrm{sp^2}$ orbitals for the $\ce{C-H}$ bonds.
The bottom line is that every model that we use in chemistry has a range of validity, and we should be careful not to use a model in a context where it is not valid. Hybridisation theory is not valid in the context of transition metal complexes, and should not be used as a means of explaining their structure, bonding, and properties.
{kind=link}