The two features you cite — trans-effect and spectrochemical series, i.e. ligand-induced crystal field split — are based on two different phenomena, two different structures and two entirely different observations that warrant two different discussions. Thus, the probability for the two trends to be inverse is $0.5$.
Spectrochemical series
The spectrochemical series in itself is already a convolute of multiple different effects and it is defined for octahedral complexes. However, if we stick to small regions of it, we can make out trends based on general observations. Here, we are dealing with four very similar ligands which are all weak-field ligands, σ bases and π bases. To understand the effect of their π basicity and to relate them to each other, we need to take a look at the molecular orbital diagram of a typical $\ce{[ML6]}$ complex accounting for π effects. Figure 1 shows such an MO scheme.
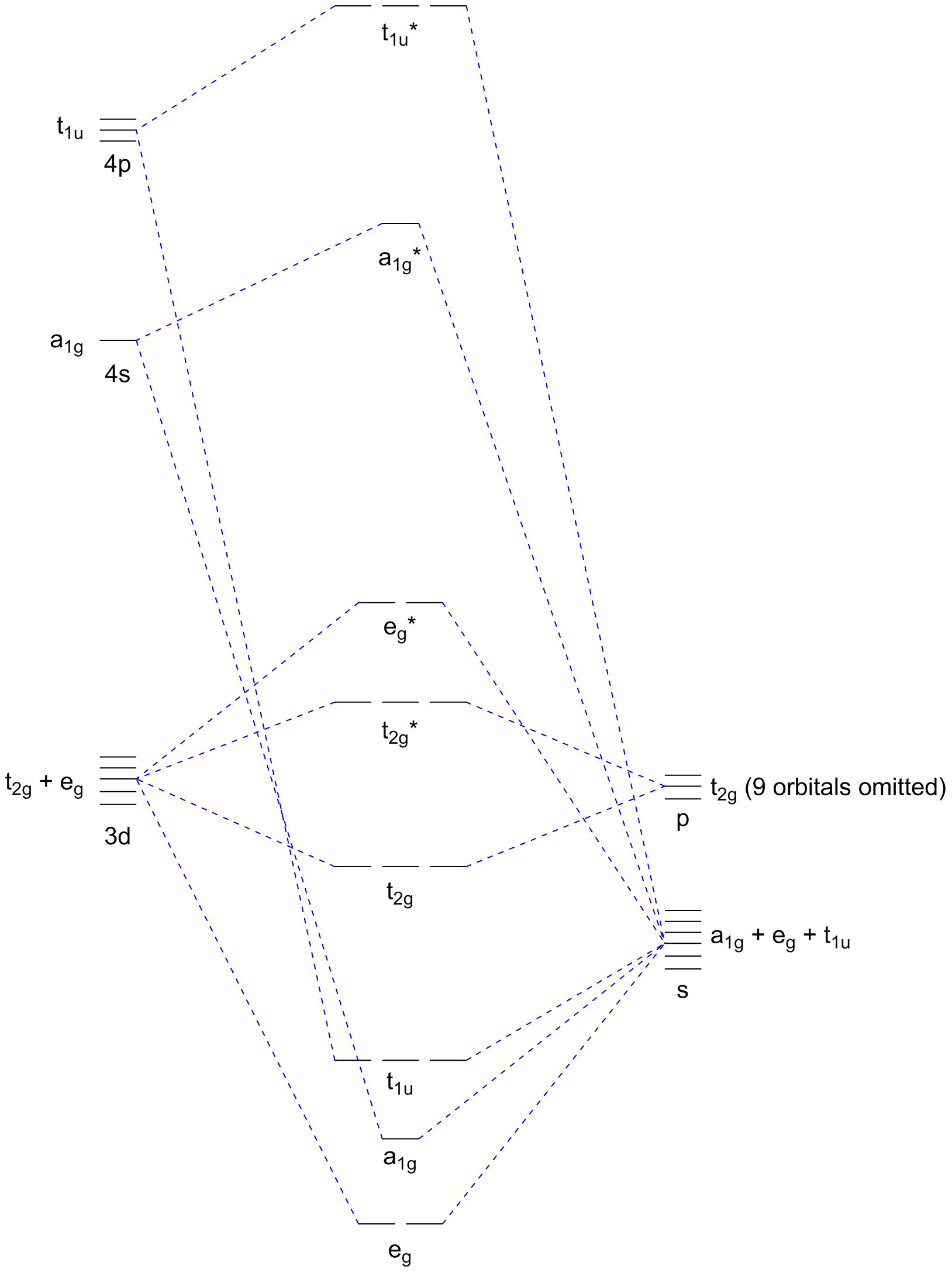
Figure 1: Octahedral $\ce{[ML6]}$ complex including σ and π ligand-metal interactions.
We see that the $\mathrm p$ orbitals (or more precisely: that $\mathrm p$ group orbital with $\mathrm{t_{2g}}$ symmetry) interact with the metal’s $\mathrm{t_{2g}}$ orbitals in a bonding/antibonding manner of π symmetry — thus the interaction is weaker than the $\mathrm{e_g}$ interaction which features σ symmetry. In the case of all halido ligands, the ligand orbitals are fully populated meaning that the metal electrons end up in the $\mathrm{t_{2g}^*}$ orbitals which have been raised with respect to the neutral (or π-effect absent) state. This reduces the energy difference between $\mathrm{t_{2g}^*}$ and $\mathrm{e_g^*}$ and thus the value of $\pu{10Dq}$ or the field split.
The different halides differ only in their electronegativity and thus in the relative positioning of their orbitals. The higher the electronegativity the lower the corresponding orbitals — i.e. fluoride will have the lowest-lying orbitals. Since these are all non-metals with relatively high ionisation energies (even for the anions that we are looking at here), they will all be below the metal orbitals. However, a larger interaction — and thereby a lower value of $\pu{10Dq}$ — is typically obtained if the interacting orbitals are close in energy.
Iodide is the one that is the least far away, meaning that the c0; interaction is largest in an $\ce{[MI6]^n-}$ complex, meaning that $\pu{10Dq}$ is lowest meaning that it is on the very weak end of the spectrochemical series. The higher up we go through the group, the lower the halide’s $\mathrm p$ orbital energies will be, thus the weaker the interaction and the higher $\pu{10Dq}$. Naturally, the orbital energy decreases monotonously and therefore the ligand series of the halides will be as observed.
Trans-effect
The trans-effect is typically observed for complexes of platinum or related elements and is typically referred to for square planar geometries. It concerns ligand exchange which has been shown by a number of experiments to proceed via an associative mechanism:
$$\ce{[MX4]^n- + L -> [MX4L]^n- -> [MX3L]^${(n-1)-}$ + X-}\tag{1}$$
The initial and final complexes are square planar, the intermediate is trigonal bipyramidal. Going by the principle of least movement, the additional ligand $\ce L$ should approach via the distinguished axis (I’m calling it $x$) towards the inital complex and displace two ligands which will now simultaneously occupy the $xy$ plane (meaning that $z$ is the axis of the two non-moving ligands). Then, one of the three $xy$ ligands will get displaced; it has to be the one that is trans to the ligand which exercises a trans-effect and it has to be one of the two in the $xy$ plane — i.e. those that were originally on the $y$ axis.
Since the pentacoordinated transition state is obviously most important here, we should draw up its molecular orbital scheme. This is a scheme much less known than the octahedral scheme shown above so I will point out a few things. The trigonal bipyramidal complex’ point group is $D_\mathrm{3h}$. The metal’s $\mathrm d$ orbitals transform as $\mathrm{a_1' + e' + e''}$ with the orbitals within each irreducible representation being $\mathrm{d}_{z^2}, (\mathrm{d}_{xy}, \mathrm{d}_{x^2-y^2})$ and $(\mathrm{d}_{xz}, \mathrm{d}_{yz})$, respectively. The ligands’ σ-symmetric orbitals transform as $\mathrm{2a_1' + e' + a_2''}$. Broken down, the $z$ ligands are $\mathrm{a_1' + a_2''}$ and the $xy$ ones are $\mathrm{a_1' + e'}$. The π-symmetric $\mathrm p$ orbitals of the $z$-ligands should transform as $\mathrm{e' + e''}$ and the π-symmetric $\mathrm p$ orbitals of the ligands in the $xy$ plane are $\mathrm{a_2' + a_2'' + e' + e''}$. We can see a lot of $\mathrm e$ irreducibles which is where all the fun takes place. For completeness, the central metal’s $\mathrm s$ orbital transforms as $\mathrm{a_1'}$ and its $\mathrm p$ orbitals as $\mathrm{a_2'' + e'}$.
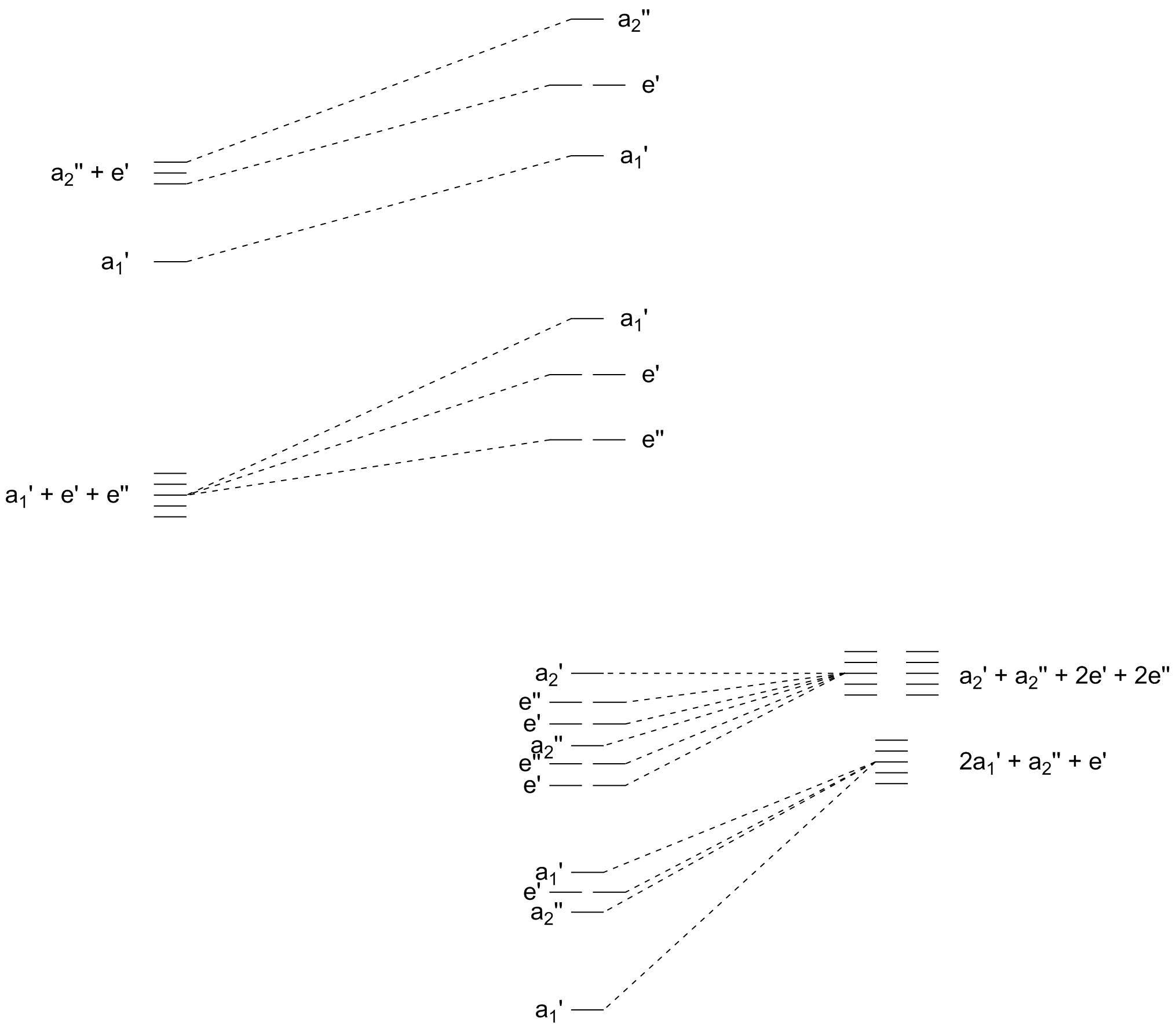
Figure 2: Qualitative molecular orbital scheme of a trigonal-bipyramidal $\ce{[ML5]}$ complex including both σ and π interactions.
The MO scheme is given in figure 2 above. The key to the analysis is to find out why certain ligands preferentially occupy the $xy$ plane but are not displaced themselves. The key lies within the orbitals that are labelled $\mathrm{e'}$: they bond in a σ fashion will all equatorial ligands in the intermediate being degenerate while in the reactant and product state they have different energies — one corresponding to a σ bond, one to a π bond. Thus, in moving from the reactant to the intermediate, we are generating a new interaction of σ symmetry which — again according to symmetry — affects all three equatorial ligands.
As I pointed out before, the lower a halide’s electronegativity the higher its orbitals energy levels and thus the greater the interaction. Therefore, by having the less electronegative halide participate in the new σ bond (i.e. by having it as part of the equatorial ligand sphere and thus by displacing the ligand trans with respect to it) will generate a more stable intermediate and will thus be a more favourable path.
Thus it is the same property of the halogens that create the two line-ups but the final effect is opposite in the two cases.