I don't know if you can do it with Avogadro, since I have no extended experience with the program. As far as I can see, there is no possibility to edit bond lengths/ angles/ dihedral angles directly. It provides a cartesian coordinate editor, but that's as tedious to manipulate your structure with as it is by doing it by hand.
I assume your problem is quite simple to be solved with ChemCraft. This program provides the tool to merge and align two structures from two different windows. This allows you to easily combine pre-computed fragments without loosing symmetry and not doing any calculations by hand.
Since I have no access to any of your structures, I am going to run you through it with an example. I am going to cup a $\ce{C70}$ nano-tube fragment with a $\ce{C40}$ buckminster fullerene, twice. Both fragments have (at least) $C_5$ symmentry, but this is no necessity. At the end we will produce a Fullerene with $D_\mathrm{5h}$ symmetry. For reasons of spacing this post, I'll include the coordinates at the end. (I am using build 485 currently, but the updated versions should still be able to perform this.)
You start with opening two instances of ChemCraft and loading each fragment into one. You want the parts to be aligned facing each other. In my layout I am modifying the $\ce{C70}$ fragment on the left, while the window on the right provides me with the fragments I use (not that it matters in this context). After this is done, you select the atoms you want to align in each window. The more atoms you select, the more input you provide for the program to align structures. Your display should look somewhat like below:
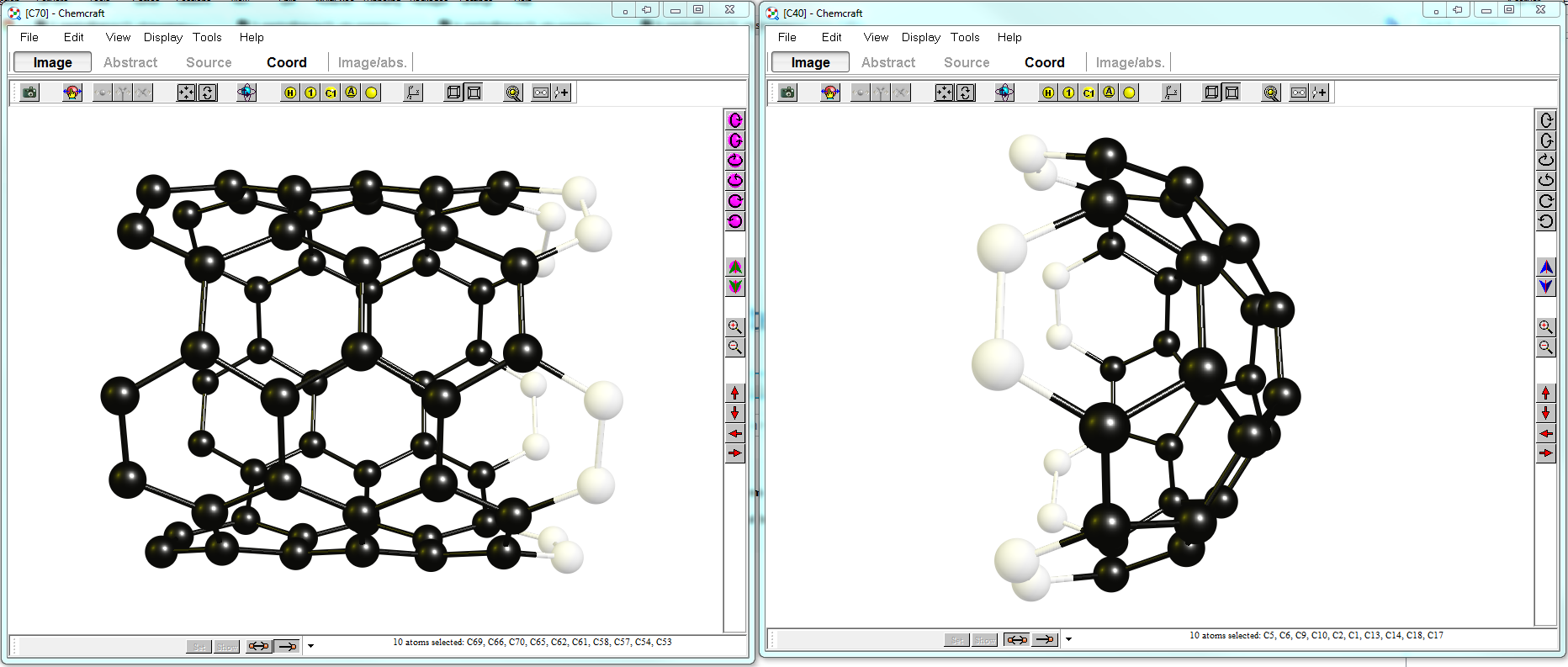
In the left window you choose "Tools > Structure combiner > Merge two structures from two Chemcraft windows by selected subunits". Because of the high symmetry the algorithm might get the orientation wrong. It is then best to try selecting (one or two) fewer atoms.
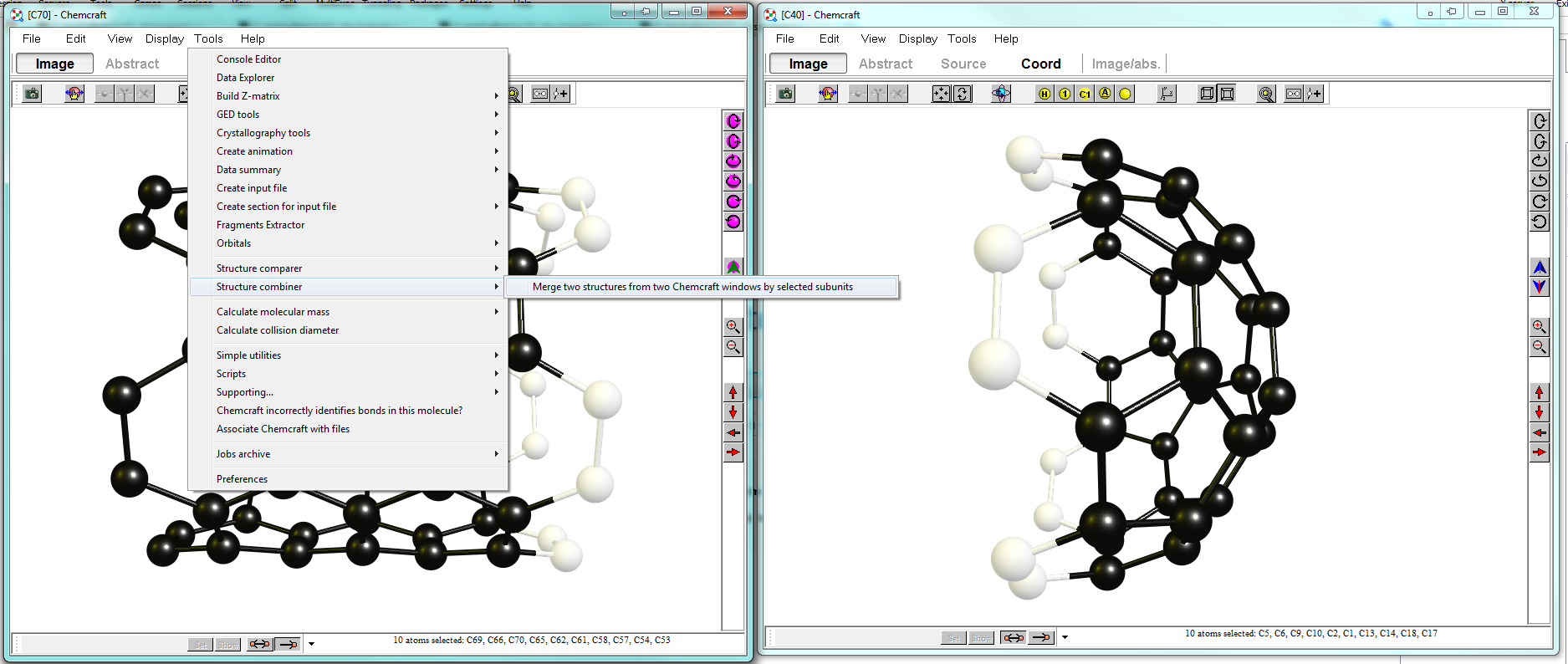
The right window should now look like below. Obviously the atoms you have taken to align are double in the mix. In some structures you can easily select them and delete them, when they are matching too well, you can switch between displays.
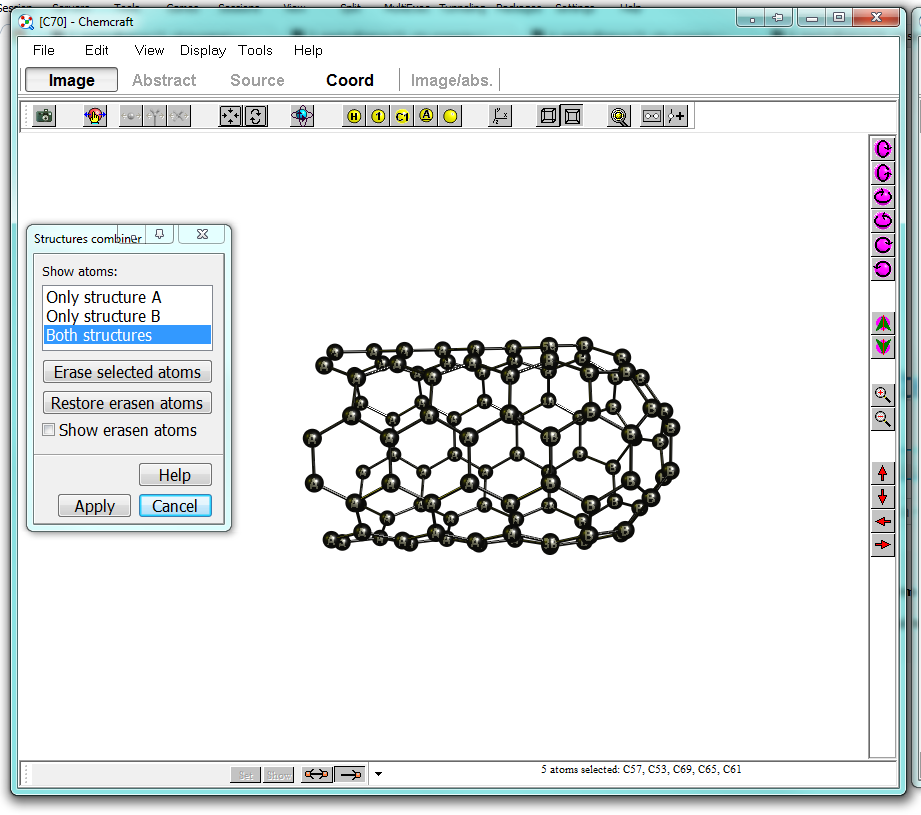
If you decide to "show erasen atoms" they will be displayed as dummies; after you click apply those will disappear and the program will re-bond you molecule.
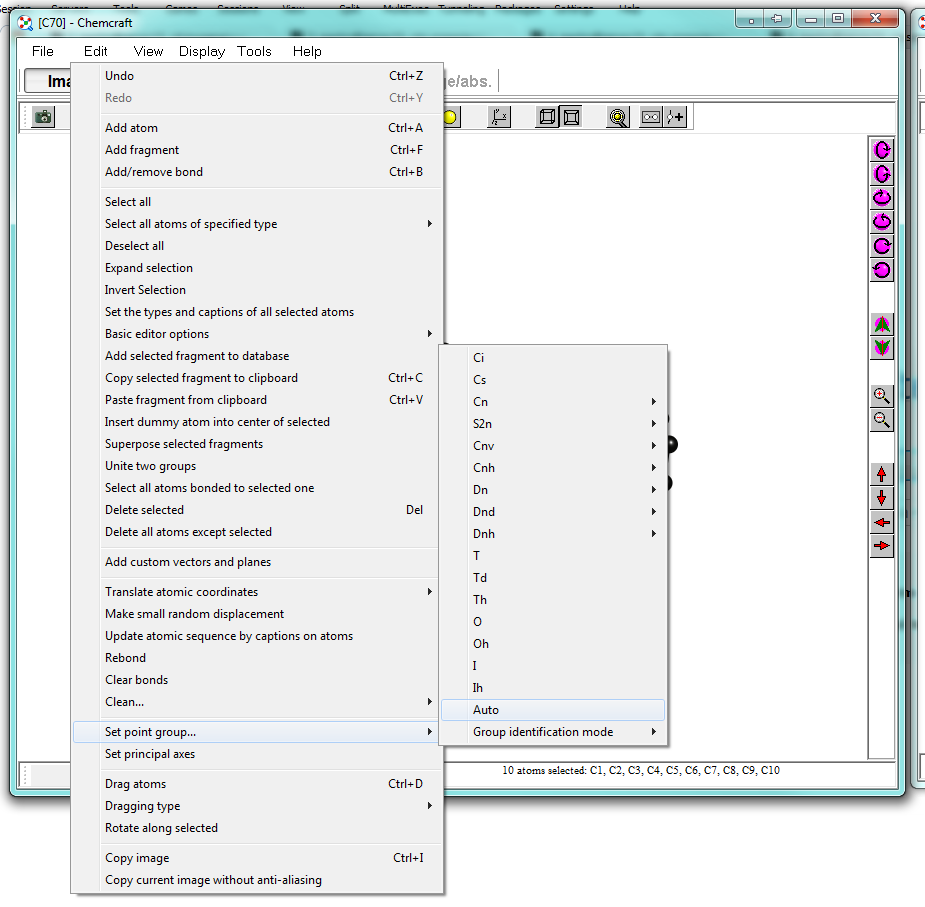
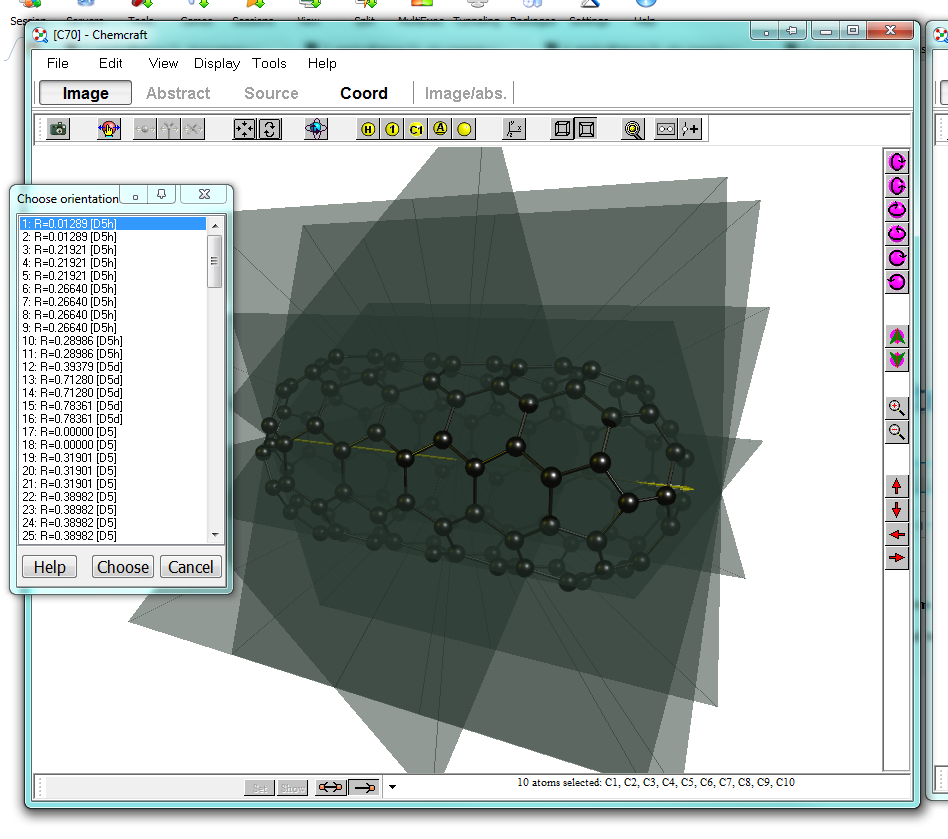
Now one side is successfully capped, and I'll proceed with adding the second cup. We have created a nice little nano-pill. From the menu, you can choose to symmetrise the molecule. Ideally in this case it should already be very, very close to $D_\mathrm{5h}$. Cartesian space will obviously introduce some artefacts. Coose "Edit > Set point group ... > Auto" to let the program determine it for you.
Indeed there is a slight variation for the point group. Fortunately the program can also correct for it.
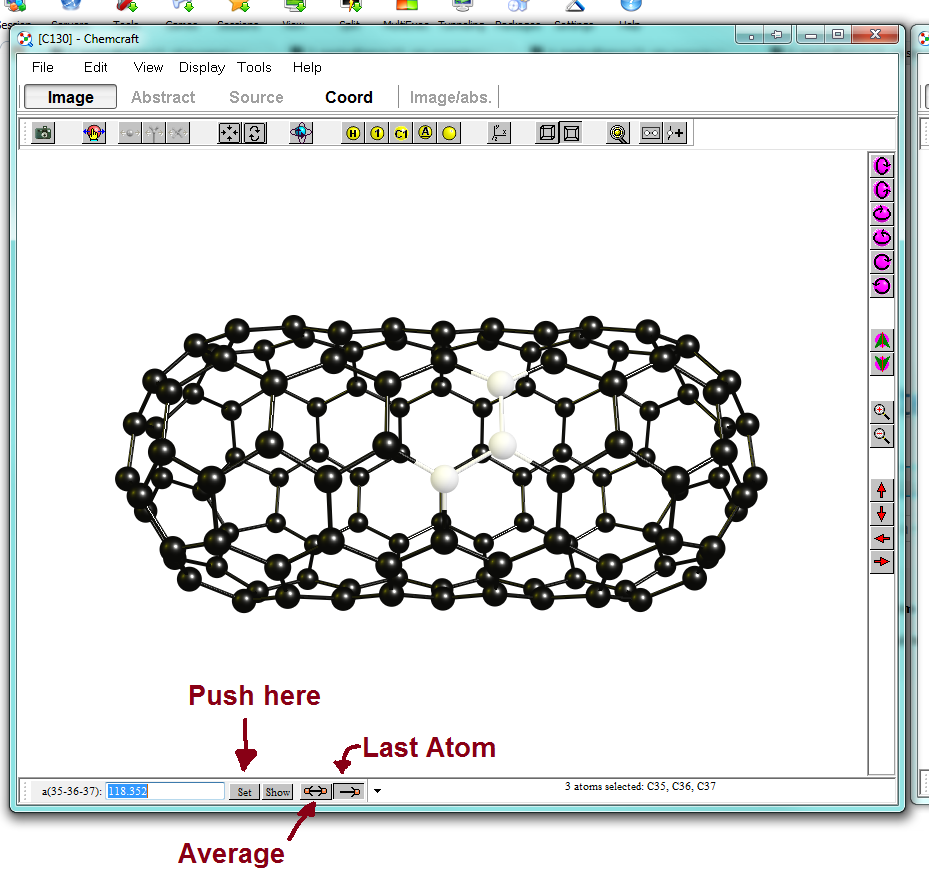
You don't need to stop there. ChameCraft comes with a xyz/ distance/ angle/ dihedral editor on the lower left corner of the window, dpending on how many atoms you select. You can choose whether you want to average the structure or only move the last selected atom. If the atoms are not connected with visible bonds, it will manipulate the whole fragment accordingly. With this feature you can fine tune everything, but then you might need your pen, paper and calculator again.
As I don't know the context of your research, it should suffice to say that these are techniques for creating suitable input geometries for more advanced calculation programs, of which there are quite a few. A well designed geometry might take crucial optimisation time off your hands. I am not sure how well suited force field methods are for nanotubes, and while Avogadro provides methodology for simple organic molecules, projects like this might just be too complex for it to be reasonable. As far as I know, Avogadro has no symmetry recognition.
Appendix
The $\ce{C70}$ fragment has the following coordinates (in angstrom):
70
symmetry c1
C 1.385307752 -3.115487343 -5.540304632
C 2.534920901 -2.280244499 -5.540304632
C 3.391088177 0.354767430 -5.540304632
C 2.951975027 1.706218740 -5.540304632
C 0.710500000 3.334745672 -5.540304632
C -0.710500000 3.334745672 -5.540304632
C -2.951975027 1.706218740 -5.540304632
C -3.391088176 0.354767430 -5.540304632
C -2.534920901 -2.280244499 -5.540304632
C -1.385307752 -3.115487343 -5.540304632
C -0.710500000 -3.334745672 -4.309125825
C 0.710500000 -3.334745672 -4.309125825
C 1.385307752 -3.115487343 -3.077947018
C 2.534920901 -2.280244499 -3.077947018
C 2.951975027 -1.706218740 -4.309125825
C 3.391088177 -0.354767430 -4.309125825
C 3.391088177 0.354767430 -3.077947018
C 2.951975027 1.706218740 -3.077947018
C 2.534920901 2.280244499 -4.309125825
C 1.385307752 3.115487343 -4.309125825
C 0.710500000 3.334745672 -3.077947018
C -0.710500000 3.334745672 -3.077947018
C -1.385307752 3.115487343 -4.309125825
C -2.534920901 2.280244499 -4.309125825
C -2.951975027 1.706218740 -3.077947018
C -3.391088176 0.354767430 -3.077947018
C -3.391088176 -0.354767430 -4.309125825
C -2.951975027 -1.706218740 -4.309125825
C -2.534920901 -2.280244499 -3.077947018
C -1.385307752 -3.115487343 -3.077947018
C -0.710500000 -3.334745672 -1.846768211
C 0.710500000 -3.334745672 -1.846768211
C 1.385307752 -3.115487343 -0.615589404
C 2.534920901 -2.280244499 -0.615589404
C 2.951975027 -1.706218740 -1.846768211
C 3.391088176 -0.354767430 -1.846768211
C 3.391088176 0.354767430 -0.615589404
C 2.951975027 1.706218740 -0.615589404
C 2.534920901 2.280244499 -1.846768211
C 1.385307752 3.115487343 -1.846768211
C 0.710500000 3.334745672 -0.615589404
C -0.710500000 3.334745672 -0.615589404
C -1.385307752 3.115487343 -1.846768211
C -2.534920901 2.280244499 -1.846768211
C -2.951975027 1.706218740 -0.615589404
C -3.391088176 0.354767430 -0.615589404
C -3.391088176 -0.354767430 -1.846768211
C -2.951975027 -1.706218740 -1.846768211
C -2.534920901 -2.280244499 -0.615589404
C -1.385307752 -3.115487343 -0.615589404
C -0.710500000 -3.334745672 0.615589404
C 0.710500000 -3.334745672 0.615589404
C 1.385307752 -3.115487343 1.846768211
C 2.534920901 -2.280244499 1.846768211
C 2.951975027 -1.706218740 0.615589404
C 3.391088176 -0.354767430 0.615589404
C 3.391088176 0.354767430 1.846768211
C 2.951975027 1.706218740 1.846768211
C 2.534920901 2.280244499 0.615589404
C 1.385307752 3.115487343 0.615589404
C 0.710500000 3.334745672 1.846768211
C -0.710500000 3.334745672 1.846768211
C -1.385307752 3.115487343 0.615589404
C -2.534920901 2.280244499 0.615589404
C -2.951975027 1.706218740 1.846768211
C -3.391088176 0.354767430 1.846768211
C -3.391088176 -0.354767430 0.615589404
C -2.951975027 -1.706218740 0.615589404
C -2.534920901 -2.280244499 1.846768211
C -1.385307752 -3.115487343 1.846768211
The $\ce{C40}$ fragment has the following coordinates:
40
symmetry c1
C -0.004840185 -3.437248260 -1.231937277
C -1.267748660 -3.000481413 -1.692390182
C -2.250219438 -2.561103110 -0.763208689
C -3.030739803 -1.417632648 -1.047784233
C -2.833456162 -0.706703611 -2.263242764
C -2.833456162 0.706703611 -2.263242764
C -3.030739803 1.417632648 -1.047784233
C -2.250219438 2.561103110 -0.763208689
C -1.267748660 3.000481413 -1.692390182
C -0.004840185 3.437248260 -1.231937277
C 0.283148522 3.437248269 0.160450194
C 1.546056995 3.000481414 0.620903095
C 2.528527766 2.561103104 -0.308278399
C 3.309048125 1.417632641 -0.023702855
C 3.111764498 0.706703610 1.191755677
C 3.111764498 -0.706703610 1.191755677
C 3.309048125 -1.417632641 -0.023702855
C 2.528527766 -2.561103104 -0.308278399
C 1.546056995 -3.000481414 0.620903095
C 0.283148522 -3.437248269 0.160450194
C -0.693482914 -3.000481414 1.084109073
C -1.963942392 -2.561103104 0.620903094
C -2.567533811 -1.417632641 1.191755675
C -3.226850870 -0.710929034 0.160450192
C -3.226850870 0.710929034 0.160450192
C -2.567533811 1.417632641 1.191755675
C -1.963942392 2.561103104 0.620903094
C -0.693482914 3.000481414 1.084109073
C -0.034165862 2.293777797 2.115414553
C 1.349945920 2.293777797 1.829137515
C 2.135133060 1.143470461 2.115414554
C 1.531541637 0.000000000 2.686267134
C 2.135133060 -1.143470461 2.115414554
C 1.349945920 -2.293777797 1.829137515
C -0.034165862 -2.293777797 2.115414553
C -0.641366192 -1.143470461 2.689680299
C -1.904274667 -0.706703610 2.229227401
C -1.904274667 0.706703610 2.229227401
C -0.641366192 1.143470461 2.689680299
C 0.139154169 0.000000000 2.974255840
Final $\ce{C130}$ nano-pill:
130
symmetry d5h
C 3.334745673 -0.710500000 -2.462357615
C 3.334745673 0.710500000 -2.462357615
C 3.115487344 1.385307752 -1.231178807
C 2.280244500 2.534920901 -1.231178807
C 1.706218741 2.951975027 -2.462357615
C 0.354767431 3.391088176 -2.462357615
C -0.354767429 3.391088176 -1.231178807
C -1.706218739 2.951975027 -1.231178807
C -2.280244498 2.534920901 -2.462357615
C -3.115487342 1.385307752 -2.462357615
C -3.334745671 0.710500000 -1.231178807
C -3.334745671 -0.710500000 -1.231178807
C -3.115487342 -1.385307752 -2.462357615
C -2.280244498 -2.534920901 -2.462357615
C -1.706218739 -2.951975027 -1.231178807
C -0.354767429 -3.391088176 -1.231178807
C 0.354767431 -3.391088176 -2.462357615
C 1.706218741 -2.951975027 -2.462357615
C 2.280244500 -2.534920901 -1.231178807
C 3.115487344 -1.385307752 -1.231178807
C 3.334745673 -0.710500000 0.000000000
C 3.334745673 0.710500000 0.000000000
C 3.115487344 1.385307752 1.231178807
C 2.280244500 2.534920901 1.231178807
C 1.706218741 2.951975027 0.000000000
C 0.354767431 3.391088176 0.000000000
C -0.354767429 3.391088176 1.231178807
C -1.706218739 2.951975027 1.231178807
C -2.280244498 2.534920901 0.000000000
C -3.115487342 1.385307752 0.000000000
C -3.334745671 0.710500000 1.231178807
C -3.334745671 -0.710500000 1.231178807
C -3.115487342 -1.385307752 0.000000000
C -2.280244498 -2.534920901 0.000000000
C -1.706218739 -2.951975027 1.231178807
C -0.354767429 -3.391088176 1.231178807
C 0.354767431 -3.391088176 0.000000000
C 1.706218741 -2.951975027 0.000000000
C 2.280244500 -2.534920901 1.231178807
C 3.115487344 -1.385307752 1.231178807
C 3.334745673 -0.710500000 2.462357615
C 3.334745673 0.710500000 2.462357615
C 1.706218741 2.951975027 2.462357615
C 0.354767431 3.391088176 2.462357615
C -2.280244498 2.534920901 2.462357615
C -3.115487342 1.385307752 2.462357615
C -3.115487342 -1.385307752 2.462357615
C -2.280244498 -2.534920901 2.462357615
C 0.354767431 -3.391088176 2.462357615
C 1.706218741 -2.951975027 2.462357615
C -1.717973404 3.000456514 3.693536421
C -0.373754410 3.437219745 3.693536421
C 0.373754411 3.437219749 4.903041136
C 1.717973407 3.000456525 4.903041139
C 2.322720745 2.561081857 3.693536424
C 3.153493774 1.417620883 3.693536424
C 3.384486708 0.706697748 4.903041140
C 3.384486708 -0.706697748 4.903041140
C 3.153493774 -1.417620883 3.693536424
C 2.322720745 -2.561081857 3.693536424
C 1.717973407 -3.000456525 4.903041139
C 0.373754411 -3.437219749 4.903041136
C -0.373754410 -3.437219745 3.693536421
C -1.717973404 -3.000456514 3.693536421
C -2.322720749 -2.561081859 4.903041134
C -3.153493775 -1.417620884 4.903041132
C -3.384486697 -0.706697747 3.693536419
C -3.384486697 0.706697747 3.693536419
C -3.153493775 1.417620884 4.903041132
C -2.322720749 2.561081859 4.903041134
C -1.579654762 2.561081856 6.105357127
C -0.227398602 3.000456514 6.105357127
C 0.745287399 2.293758766 6.848429277
C 1.947593421 2.293758772 6.105357132
C 2.783333689 1.143460975 6.105357132
C 2.411800691 0.000000000 6.848429279
C 2.783333689 -1.143460975 6.105357132
C 1.947593421 -2.293758772 6.105357132
C 0.745287399 -2.293758766 6.848429277
C -0.227398602 -3.000456514 6.105357127
C -1.579654762 -2.561081857 6.105357127
C -1.951187752 -1.417620880 6.848429274
C -2.923873754 -0.710923134 6.105357124
C -2.923873754 0.710923134 6.105357124
C -1.951187752 1.417620880 6.848429274
C -0.972686005 0.706697746 7.595944297
C 0.371532989 1.143460974 7.595944302
C 1.202306016 0.000000000 7.595944306
C 0.371532989 -1.143460974 7.595944302
C -0.972686005 -0.706697746 7.595944297
C -1.717973404 -3.000456514 -3.693536421
C -0.373754410 -3.437219745 -3.693536421
C 0.373754411 -3.437219749 -4.903041136
C 1.717973407 -3.000456525 -4.903041139
C 2.322720745 -2.561081857 -3.693536424
C 3.153493774 -1.417620883 -3.693536424
C 3.384486708 -0.706697748 -4.903041140
C 3.384486708 0.706697748 -4.903041140
C 3.153493774 1.417620883 -3.693536424
C 2.322720745 2.561081857 -3.693536424
C 1.717973407 3.000456525 -4.903041139
C 0.373754411 3.437219749 -4.903041136
C -0.373754410 3.437219745 -3.693536421
C -1.717973404 3.000456514 -3.693536421
C -2.322720749 2.561081859 -4.903041134
C -3.153493775 1.417620884 -4.903041132
C -3.384486697 0.706697747 -3.693536419
C -3.384486697 -0.706697747 -3.693536419
C -3.153493775 -1.417620884 -4.903041132
C -2.322720749 -2.561081859 -4.903041134
C -1.579654762 -2.561081857 -6.105357127
C -0.227398602 -3.000456514 -6.105357127
C 0.745287399 -2.293758766 -6.848429277
C 1.947593421 -2.293758772 -6.105357132
C 2.783333689 -1.143460975 -6.105357132
C 2.411800691 0.000000000 -6.848429279
C 2.783333689 1.143460975 -6.105357132
C 1.947593421 2.293758772 -6.105357132
C 0.745287399 2.293758766 -6.848429277
C -0.227398602 3.000456514 -6.105357127
C -1.579654762 2.561081856 -6.105357127
C -1.951187752 1.417620880 -6.848429274
C -2.923873754 0.710923134 -6.105357124
C -2.923873754 -0.710923134 -6.105357124
C -1.951187752 -1.417620880 -6.848429274
C -0.972686005 -0.706697746 -7.595944297
C 0.371532989 -1.143460974 -7.595944302
C 1.202306016 0.000000000 -7.595944306
C 0.371532989 1.143460974 -7.595944302
C -0.972686005 0.706697746 -7.595944297