According to Noack and Jones, unsaturated ketones with an α substituent — even if it is only methyl — generally adopt an s-trans conformation due to steric reasons. Only if the α substituent is hydrogen can an equilibrium between s-cis and s-trans be observed.[1] Therefore, we should better draw the structure this way:
$\hspace{30ex}$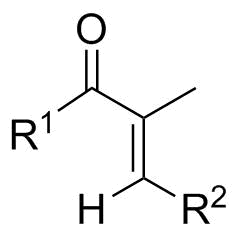

$\hspace{28ex}$s-trans configuration of α-methylenones
I already explicitly added the unsaturated hydrogen atom to the image. As we can see, it is either rather close to the keto group or pointing away from it. If the alkene’s configuration is (E) and the hydrogen is thus pointing ‘inward’ it experiences a larger deshielding due to the close carbonyl group. Therefore, its signal in 1H-NMR is shifted to lower field when compared to the (Z)-configured double bond.
You can also compare experimental shifts. Unfortunately I didn’t find a pair of identical molecules differing only in the double bond orientation, and not even the solvent is the same, but I think the data still shows a clear trend, namely that for (E) enones the chemical shift is in the high alkene range while for the (Z) enones it is in the low range:
$\hspace{33ex}$
(E)-3-methylhex-3-en-2-one. 1H-NMR ($500~\mathrm{MHz}$, $\ce{CDCl3}$) $\delta =$ $\mathbf{6.59}$ (tq, $J = 7.2,1.3~\mathrm{Hz}$, 1 H), $2.28$ (s, 3 H), $2.24$ (app. qnt, $J = 7.4~\mathrm{Hz}$, 2 H), $1.73$ (s, 3 H), $1.05$ (t, $J = 7.6~\mathrm{Hz}$, 3 H).[2]
$\hspace{33ex}$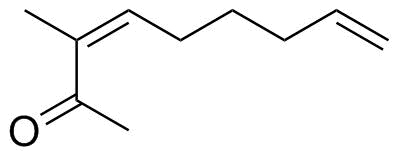
(Z)-3-methylnona-3,8-dien-2-one. 1H-NMR ($300~\mathrm{MHz}$, $\ce{C6D6}$) $\delta =$ $5.71$ (ddt, $J = 17.1, 10.5, 6.6~\mathrm{Hz}$, 1H), $\mathbf{5.38}$ (tt, $J = 7.5, 1.5~\mathrm{Hz}$, 1 H), $4.92$–$5.02$ (m, 2 H), $2.29$ (q, $J = 7.5~\mathrm{Hz}$, 2 H), $1.92$ (q, $J = 7.2~\mathrm{Hz}$, 2 H), $1.85$ (s, 3 H), $1.60$ (t, $J = 2.7~\mathrm{Hz}$, 3 H), $1.35$ (qnt, $J = 7.5~\mathrm{Hz}$, 2 H).[3]
References:
[1]: K. Noack, R. N. Jones, Can. J. Chem. 1961, 39, 2225. DOI: 10.1139/v61-294.
[2]: W. F. Austin, Y. Zhang, R. L. Danheiser, Org. Lett. 2005, 7, 3905. DOI: 10.1021/ol051307b.
[3]: L. Jiao, C. Yuan, Z.-X. Yu, J. Am. Chem. Soc. 2008, 130, 4421. DOI: 10.1021/ja7100449.
