As Mad Scientist said in the comments, measurement of the transverse signal in NMR occurs in both the $x$- and $y$-directions. This is known as quadrature detection. In practice this is not achieved with two detector coils, but rather with a "trick" which is described more fully in Keeler's Understanding NMR Spectroscopy, 1st ed., section 5.6. For a more mathematical description, see also Appendix A.5 of Levitt's Spin Dynamics, 2nd ed.
Instead of having two separate quantities $M_x$ and $M_y$, it's easier to lump them into one complex quantity $M$:
$$M = M_x + \mathrm{i}M_y$$
For example, if we only have one spin with equilibrium magnetisation $M_0$ pointing along the $z$-axis, then following a $90^\circ (y)$ pulse (which rotates the magnetisation vector to $x$),
$$\begin{align}
M_x &= M_0 & M_y = 0
\end{align}$$
and after a period of free precession $t$
$$\begin{align}
M_x &= M_0\cos(\Omega t)\mathrm{e}^{-t/T_2} & M_y = M_0\sin(\Omega t)\mathrm{e}^{-t/T_2}
\end{align}$$
and $M = M_x + \mathrm{i}M_y = M_0\mathrm{e}^{\mathrm{i}\Omega t}\mathrm{e}^{-t/T_2}$, where $T_2$ is the spin-spin relaxation time. Fourier transformation of this complex signal gives a spectrum $S(\omega) = A(\omega) - \mathrm{i}D(\omega)$. There is a constant of proportionality which doesn't matter and has been dropped. The real and complex components are known as the absorption and dispersion Lorentzians respectively:
$$\begin{align}
A(\omega) &= \frac{1/T_2}{(1/T_2)^2 + (\omega - \Omega)^2} & D(\omega) = \frac{\omega - \Omega}{(1/T_2)^2 + (\omega - \Omega)^2}
\end{align}$$

(Diagram from Keeler's Understanding NMR Spectroscopy, 2nd ed. (p 84).) The absorption component is usually what is shown in the actual NMR spectrum. There are some complexities because the spectrum has to be phase shifted, but the idea holds.
For a single spin it is very easy to use product operators instead of the vector model. After that, to obtain the expressions in your question, you just generalise to multiple spins. I might come back to this some time later, but for now, this will suffice.
The benefit of quadrature recording (which seems to have been cut from Keeler's 2nd ed., but remains in the 1st ed.) is that it allows us to find out the sign of the offset $\Omega$. Let's say that instead of having quadrature recording we only measure along the $x$-axis. Our signal is
$$\begin{align}
M &= M_x \\
&= M_0\cos(\Omega t)\mathrm{e}^{-t/T_2} \\
&= \frac{1}{2}(M_0\mathrm{e}^{\mathrm{i}\Omega t}\mathrm{e}^{-t/T_2} + M_0\mathrm{e}^{-\mathrm{i}\Omega t}\mathrm{e}^{-t/T_2})
\end{align}$$
It's relatively straightforward to show that for a function $M = M(t)$, if $\mathcal{F}[M(t)] = f(\omega)$:
$$\mathcal{F}[M^*(t)] = [f(-\omega)]^*$$
and using this, we can show that if we take the Fourier transform of the one-dimensional magnetisation, we get
$$\begin{align}
S(\omega) = \frac{1}{2}[A(\omega) - \mathrm{i}D(\omega) + A(-\omega) + \mathrm{i}D(-\omega)]
\end{align}$$
and if you take the full expression for this and replace $\Omega$ by $-\Omega$, you will find that $S(\omega)$ is the same regardless of the sign of the offset $\Omega$. This means that our spin of interest could have either a positive or a negative offset, relative to the spectrometer reference frequency $\omega_\mathrm{rf}$, but we would not be able to determine it.
In theory, if you could control the phase of the signal you are detecting, single detection would allow you to extract the sign of the offset. For example, in our example above, if you measure $M_y$ instead of $M_x$, you'll find that there is a difference when you replace $\Omega$ by $-\Omega$. Here's a schematic from Gunther's NMR Spectroscopy, 3rd ed. (p 265):
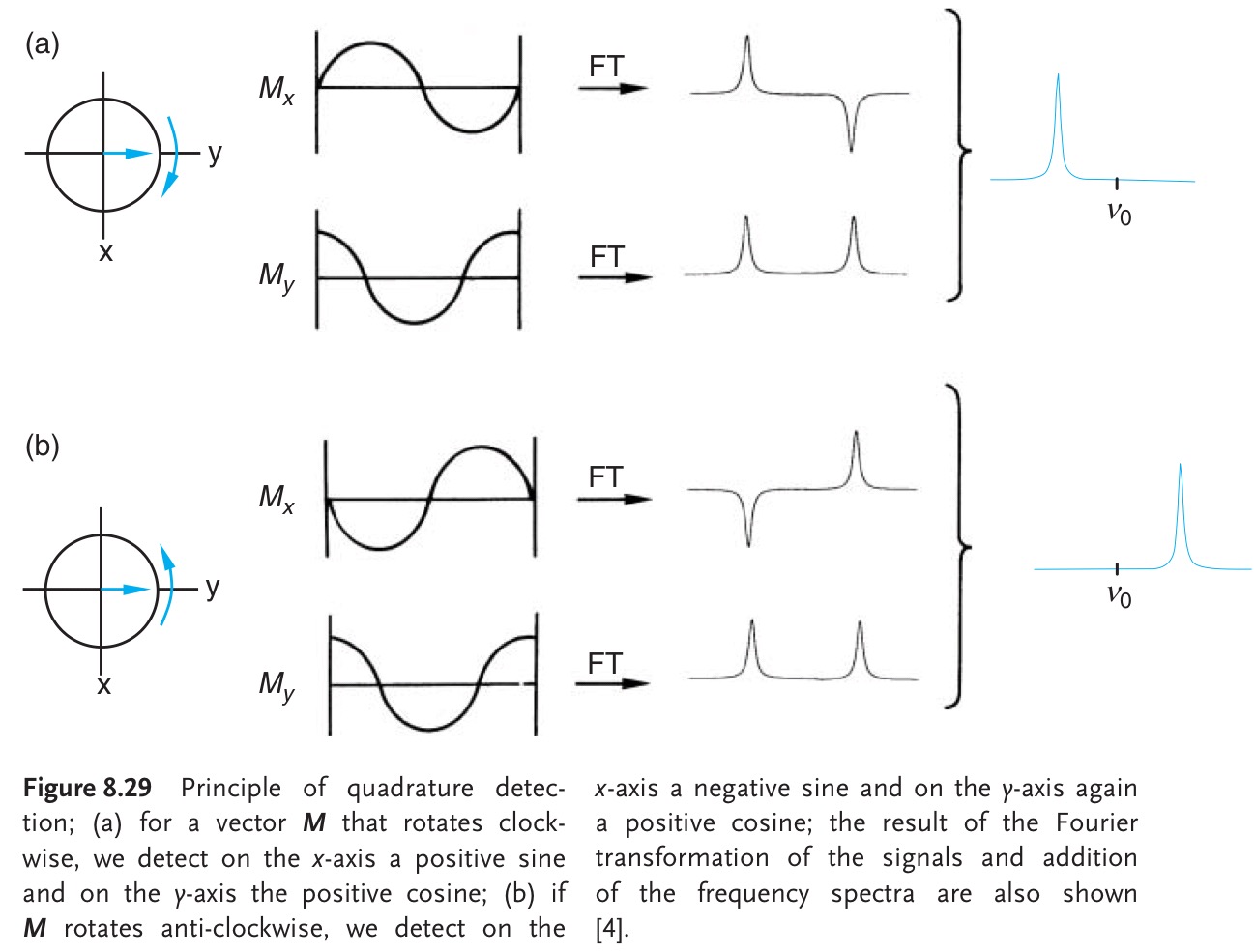
Here, the author Fourier transformed $M_x$ and $M_y$ separately before adding them up. Since the Fourier transform is linear, this has the same effect as adding $M_x$ and $M_y$ and Fourier transforming the sum. In any case, it shows that if the magnetisation vector starts off along the $y$-axis, you could, in theory, measure only the $x$-magnetisation to determine whether the offset is negative (case a) or positive (case b).
Such a spectrum would be very difficult to interpret, and would probably be impossible to analyse if positive and negative peaks overlapped. On top of that, it would also require you to know the exact phase difference between the initial signal and your single detection coil. I believe this is (due to several reasons) not experimentally possible, so unfortunately, there's no way to extract this information using single detection. (This is also part of the reason why NMR spectra have to be phase corrected – for the average NMR user the computer does this automatically.)
Now, we've established that you need quadrature detection to determine the sign of the offset. However, in the final NMR spectrum, we don't really look at offsets; we look at resonance frequencies. (To be precise, chemical shifts, but those are linearly related to resonance frequencies.) So, if you really wanted to, you could use single detection while placing the reference frequency at the far end of the spectrum. For example, if you want to measure peaks from $400.000$ to $400.001~\mathrm{MHz}$, you can place the reference frequency at $400.002~\mathrm{MHz}$. Then, by virtue of choosing an overly high value for $\omega_\mathrm{rf}$, you know that your offset $\Omega$ has to be negative.
However, a rf pulse centred at $400.002~\mathrm{MHz}$ would be less effective at exciting the spins (as it is further away from being on-resonance), and essentially you are throwing away half of the rf pulse (any components of the rf pulse with frequency greater than $400.002~\mathrm{MHz}$ are wasted). So, it's preferable to put $\omega_\mathrm{rf}$ right in the middle of the spectrum; but as said before, that would necessitate quadrature detection so that you can obtain information about the sign of the offset.
As a final comment, the fact you note about how $\hat{I}_{\!x} + \mathrm{i}\hat{I}_{\!y}$ is the raising operator $\hat{I}_{\!+}$ is reflected in many books when they discuss coherence transfer pathways: often it is said that the detected signal has coherence order $-1$. This corresponds precisely to detecting the $\hat{I}_+$ operator, or specifically, one off-diagonal entry of the density matrix.