I have multiple PDB files containing a single molecules of polysaccharides, with a single chain and a long list of all the atoms -they are not divided by residue.
The PDB files were created from ChemDraw files using OpenBabel and Corina applet form molecular-networks. The workflow was: .cdx <-> OpenBabel <-> smiles/sdf <-> Corina <-> pdb.
What i end up with is this:
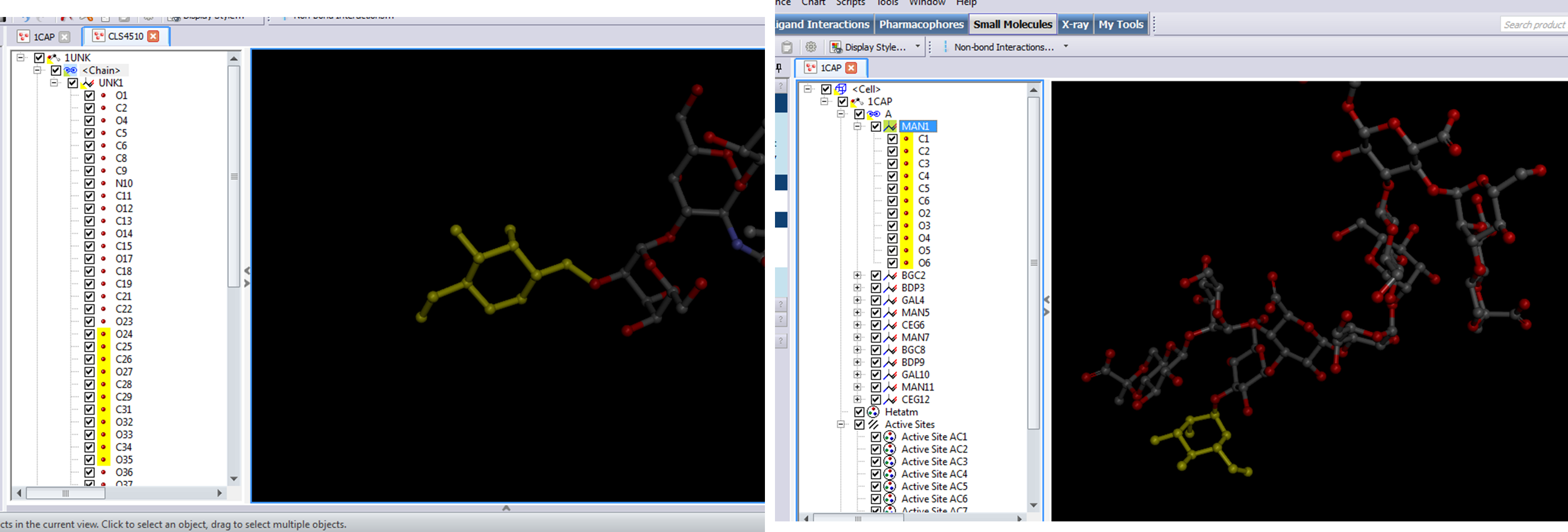
How do i easily break up the list into individual residues(individual monosaccharides)? I trying to something similar to 1CAP
This is what the PDB file looks like after CORINA:
HEADER UNK 15-05-08 1UNK
REMARK 1 corina 3.48 0000 08.02.2010
HETATM 1 C1 UNK 1 6.576 -2.524 4.463 1.00 20.00
HETATM 2 H2 UNK 1 6.573 -2.048 5.444 1.00 20.00
HETATM 3 C3 UNK 1 5.876 -1.614 3.449 1.00 20.00
HETATM 4 C4 UNK 1 4.411 -1.438 3.863 1.00 20.00
HETATM 5 C5 UNK 1 3.748 -2.815 3.964 1.00 20.00
HETATM 6 H6 UNK 1 3.760 -3.296 2.986 1.00 20.00
HETATM 7 O7 UNK 1 4.467 -3.621 4.900 1.00 20.00
HETATM 8 C8 UNK 1 5.829 -3.859 4.541 1.00 20.00
HETATM 9 C9 UNK 1 6.487 -4.751 5.595 1.00 20.00
HETATM 10 O10 UNK 1 5.864 -6.037 5.587 1.00 20.00
HETATM 11 O11 UNK 1 2.398 -2.662 4.404 1.00 20.00
HETATM 12 C12 UNK 1 1.655 -3.882 4.435 1.00 20.00
HETATM 13 C13 UNK 1 0.198 -3.584 4.795 1.00 20.00
HETATM 14 C14 UNK 1 -0.566 -4.899 4.964 1.00 20.00
HETATM 15 H15 UNK 1 -0.496 -5.481 4.045 1.00 20.00
HETATM 16 C16 UNK 1 -2.036 -4.592 5.265 1.00 20.00
HETATM 17 C17 UNK 1 -2.607 -3.729 4.136 1.00 20.00
HETATM 18 C18 UNK 1 -1.764 -2.458 4.000 1.00 20.00
...
...
HETATM 286 H UNK 1 14.974 11.280 1.314 1.00 20.00
HETATM 287 H UNK 1 15.417 11.215 -0.409 1.00 20.00
HETATM 288 H UNK 1 15.300 13.559 0.392 1.00 20.00
HETATM 289 H UNK 1 14.052 13.241 -0.836 1.00 20.00
HETATM 290 H UNK 1 13.609 13.306 0.886 1.00 20.00
HETATM 291 H UNK 1 9.253 -4.179 6.144 1.00 20.00
HETATM 292 H UNK 1 11.359 -2.412 4.593 1.00 20.00
CONECT 1 2 8 3 115
CONECT 2 1
CONECT 3 1 4 86 120
CONECT 4 3 5 69 121
CONECT 5 4 6 7 11
CONECT 6 5
CONECT 7 5 8
CONECT 8 7 1 9 122
...
CONECT 289 114
CONECT 290 114
CONECT 291 118
CONECT 292 119
END