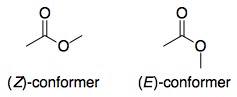
Overview
The only interesting thing about lactones is that if the ring size is relatively small, they necessarily adopt the (E) conformation. Therefore, the question essentially boils down to: for a generic ester, why is the (E)-conformer 1 more acidic than the (Z)-conformer 2? This is a question that is easier to investigate, since we remove all other variables e.g. molecular structure.
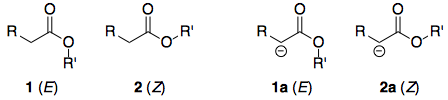
Before diving into the details, I'll state the answer: 2 is more stable than 1. Likewise, if one compares the enolate anions, 2a is more stable than 1a, but the difference in stability between the anions is not as large as in the esters. Hence, $\Delta G^\circ$ for the deprotonation of 2 is larger than that for the deprotonation of 1, and 2 is less acidic. Schematically:
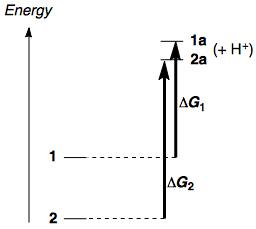
In physical organic chemistry the difference between the two values of $\Delta G$ is referred to as $\Delta \Delta G$. In this case, we can define $\Delta G_2 - \Delta G_1 \equiv \Delta \Delta G$, which we expect to be positive.
Neutral molecules
The two conformations of esters have been studied for quite a long time; as far as I can tell there seem to be two major reasons put forth as for why 2 is more stable than 1.
In the literature, the accepted argument seems to be based on a minimisation of dipole-dipole repulsions in 2.1–4 In particular, Roos et al.2 investigated the three possible contributing factors of (1) dipole-dipole repulsions, (2) repulsions between lone pairs on oxygens, and (3) steric repulsions between R and R'. Their conclusion was that the dipole-dipole repulsions were the most significant, and this analysis seems to have been accepted by later authors.
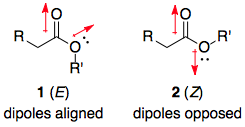
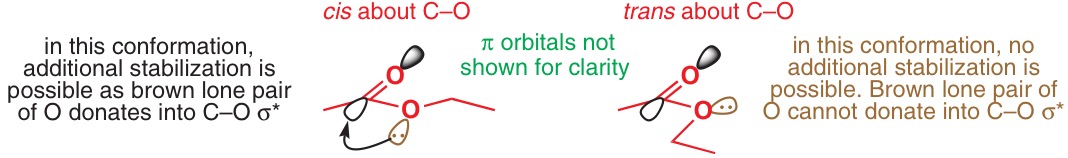
In textbooks, however, the n-to-σ* anomeric donation seems to prevail.5–9 However, no literature references are given, and Deslongchamps6 admits that "there is no direct experimental evidence to show the importance of [these] secondary electronic effects" (p 57). This was written in 1983, and even computational studies since then have not mentioned the existence of any such effect.
What everybody can agree on, though, is that 2 is more stable than 1. The energy difference was determined by IR spectroscopy to be $\pu{4.75 kcal mol-1}$ for methyl formate, and $\pu{8.5 kcal mol-1}$ for methyl acetate (the latter being larger because of steric repulsions between methyl groups).10 These values are corroborated by multiple computational studies.2–4
Enolates
I actually get the feeling that some of the textbooks get sloppy, or gloss over this aspect. Reading Clayden5 for example, the impression I get is that because 1 is more unstable than 2, it reacts more readily (with bases or nucleophiles). However, in comparing $\Delta G$ for two different reactions, one must be careful to look at the relative stabilities of not just the reactants, but also the products. If 2a is also more stable than 1a, and to the same extent ($\Delta\Delta G = 0$), then there is no reason to expect any difference in acidity.
The question that we now turn to is therefore: why is the relative stabilisation of 2 partially lost upon deprotonation to form 2a? The answer will, of course, depend on which of the two above explanations you subscribe to.
First, the dipole minimisation argument. Upon deprotonation, negative charge develops on the α carbon; hence as a whole, the enolate moiety has a smaller dipole moment than an undeprotonated carbonyl moiety. (Since the carbonyl oxygen bears more negative charge than the α carbon, the direction of the dipole moment is roughly the same.) Houk3 has reported this in the form of Mulliken partial charges on atoms, and Wiberg4 in the form of electron populations, but the key point is the same: when the dipole of the carbonyl group is smaller, the dipole-dipole interactions are weaker. Consequently, while 2a is still more stable than 1a, the energy difference is not as large as it is in 2 versus 1. Houk's ab initio calculations (6-31+G*//3-21G) on methyl acetate3 gives the energy difference between 2a and 1a as $\pu{3.8 kcal mol-1}$, and that between 2 and 1 as $\pu{9.2 kcal mol-1}$. Therefore, the difference $\Delta\Delta G$ is roughly $\pu{5 kcal mol-1}$.
Extra: in a later article, Houk and Jorgensen11 revisited the topic to calculate the potential effects of solvation. A polar solvent would be expected to preferentially stabilise the more polar (E)-conformers 1 and 1a (which are more polar since the dipoles reinforce each other). It turns out that solvation in either water or acetonitrile only reduces $\Delta \Delta G$ by $\pu{1 kcal mol-1}$.
As for the stereoelectronic argument, only Alabugin7 offers any possible explanation:
A number of factors can contribute to this difference but, at least partially, this effect can be explained by the reduced acceptor ability of the C–O bond of the enolate where the oxygen atom has significant negative charge and by the subsequent decrease in the energy of the nO to σ*C–O stabilisation.
Further reading
Much of the research carried out has been targeted at understanding the anomalously high acidity of Meldrum's acid, and a literature search for this will yield many of the references cited here. The $\mathrm pK_\mathrm a$ values below (DMSO) are taken from Arnett and Harrelson12:
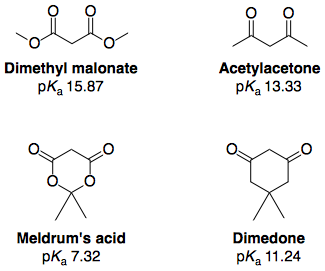
The dramatic increase in acidity upon "cyclisation" of a diester - to the point where the cyclic diester (Meldrum's acid) was more acidic than a cyclic diketone (dimedone) - was traditionally explained with the above information (i.e. it is more acidic because it has two (E)-esters).
However, newer research suggests that there is also an extra stereoelectronic factor that accounts for the unusually high acidity, namely donation from the ester oxygen lone pair into the σ* orbital of the adjacent $\ce{C(sp^3)–O}$ single bond. This stabilising effect is more pronounced in the conjugate base because the oxygen has greater electron density and the donor orbital has a higher energy.13
Notes and references
Huisgen, R. Neuere Beiträge zur Chemie mittlerer Ringe. Angew. Chem. 1957, 69 (11), 341–359. DOI: 10.1002/ange.19570691102. Disclaimer: I can't quite read German, but none of the figures indicate any sort of stereoelectronic effect.
Wennerstrom, H.; Forsen, S.; Roos, B. Ester group. I. Ab initio calculations on methyl formate. J. Phys. Chem. 1972, 76 (17), 2430–2436. DOI: 10.1021/j100661a015.
Wang, X.; Houk, K. N. Theoretical elucidation of the origin of the anomalously high acidity of Meldrum's acid. J. Am. Chem. Soc. 1988, 110 (6), 1870–1872. DOI: 10.1021/ja00214a032.
Wiberg, K. B.; Laidig, K. E. Acidity of (Z)- and (E)-methyl acetates: relationship to Meldrum's acid. J. Am. Chem. Soc. 1988, 110 (6), 1872–1874. DOI: 10.1021/ja00214a033.
Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford UP: Oxford, U.K., 2012.
Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry; Pergamon Press: Oxford, U.K., 1983.
Alabugin, I. V. Stereoelectronic Effects; Wiley: Chichester, U.K., 2016.
Kirby, A. J. Stereoelectronic Effects; Oxford UP: Oxford, U.K., 1996.
D. A. Evans' famous Chem 206 notes also only mention this stereoelectronic effect. The set of notes is downloadable here (the relevant part is in Lecture 2).
Blom, C. E.; Günthard, H. H. Rotational isomerism in methyl formate and methyl acetate; a low-temperature matrix infrared study using thermal molecular beams. Chem. Phys. Lett. 1981, 84 (2), 267–271. DOI: 10.1016/0009-2614(81)80342-9.
Evanseck, J. D.; Houk, K. N.; Briggs, J. M.; Jorgensen, W. L. Quantification of Solvent Effects on the Acidities of Z and E Esters from Fluid Simulations. J. Am. Chem. Soc. 1994, 116 (23), 10630–10638. DOI: 10.1021/ja00102a032.
Arnett, E. M.; Harrelson, J. A., Jr. Ion pairing and reactivity of enolate anions. 7. A spectacular example of the importance of rotational barriers: the ionization of Meldrum's acid. J. Am. Chem. Soc. 1987, 109 (3), 809–812. DOI: 10.1021/ja00237a028.
Byun, K.; Mo, Y.; Gao, J. New Insight on the Origin of the Unusual Acidity of Meldrum's Acid from ab Initio and Combined QM/MM Simulation Study. J. Am. Chem. Soc. 2001, 123 (17), 3974–3979. DOI: 10.1021/ja001369r.