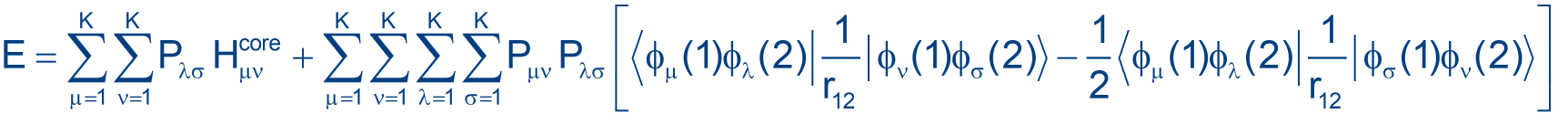In the context of Hartree-Fock theory, using Roothaan formalism, we write:
FC=SCE
where F is the matrix of the Fock operator, C are the coefficients to be used in the construction of the wave function from the basis set, S is the overlap matrix and E are the energies.
The problem is that the coefficients in C are actually included in F, if we make it explicit. Using Roothaan approach, we just need to choose a basis set to be able to compute all the integrals in F and in S . . . but we need a "density matrix" (a part of F) that takes into account what molecular orbitals are occupied, in order to (partially) account for electron-electron Coulomb interaction.
So the approach is:
- Choose basis set and molecular geometry
- Compute all the integrals, as stated above
- Guess initial density matrix
- Solve the matrix equation to find the energies E
We are guided by the variational principle that ensures we cannot "overshoot" in the search for the minimum energy . . . the lower we find, the better (=closer to true ground state energy).
If up to this point i got it right, then my question is the following; after one cycle, as described above, i'd be tempted to say that the correct procedure is this:
I) Use some kind of minimization algorithm to make a new guess for the density matrix, which should be equivalent to saying 'make a new guess for the coefficients in C' II) Repeat the cycle above III) Evaluate if the new energy minimum is below the previous one (or check for another, more convenient, convergence criterion) IV) Repeat until convergence
What instead seems to be the correct way is that, after one cycle, you use the energies to compute a new set of coefficients, which you then use to construct a new density matrix and so on . . . you are now in a cycle (SCF) and you can stop iterating once you achieve convergence according to the criteria you set.
What seems strange to me is that if you guess a density matrix, then solve the Roothaan matrix equation, i would expect the energies to be "consistent" with the guess we've made . . . meaning that we will NOT get new coefficients from the procedure. Could you give me an intuition why this is not the case? Is everything else i've stated above correct?
Thank you.