General case
There is indeed a mathematical theorem that deals with the number of nodes an eigenfunction corresponding to a certain eigenvalue can possess.
It was laid down by Courant$^{[1, 2]}$ and it states the following:
Given the self-adjoint second order (partial) differential equation
\begin{equation} \left(\hat{L} + \lambda \rho(\mathbf{x}) \right)
u(\mathbf{x}) = 0 \end{equation}
(where $\hat{L} = L(\mathbf{\Delta}, \mathbf{x})$ is a linear,
hermitian differential operator, $\rho(\mathbf{x})$ is positive and
bounded, and $\lambda$ is the eigenvalue) for a domain $G$ with
homogeneous boundary conditions, that is $u(\mathbf{x}) = 0$ on the
boundary of the region $G$; if its eigenfunctions are ordered
according to increasing eigenvalues, then the nodes of the $n^{\text{th}}$
eigenfunction divide the domain into no more than $n$ subdomains. The
nodal set of $u(\mathbf{x})$ is defined as the set of points
$\mathbf{x}$ such that $u(\mathbf{x}) = 0$. No assumptions are made
about the number of independent variables.
The proof is rather involved and so I won't show it here. But if you want you can look it up in [1] or here.
So, Courant's nodal line theorem tells us, that if we order the possible energy eigenvalues of the time-independent Schroedinger equation as $\lambda_1 \leq \lambda_2 \leq \lambda_3 \leq \dots$, then (depending on precisely how you set up the numbering) the $n^{\text{th}}$ eigenfunction, $\Psi_{n}$ (the one with energy eigenvalue $\lambda_n$) has at most $n$ nodes (including the trivial one at the boundary $\mathbf{x} \to \infty$).
Unfortunately, this gives you only an upper bound for the number of nodes a wave function with a certain energy eigenvalue may possess. So, all we know is that the ground state wave function $\Psi_{1}$ cannot have any nodes within the region $G$ (in total it has one node, namely the one at $\mathbf{x} \to \infty$). Wave functions for higher $n$ may possess up to $n-1$ nodes within $G$ but may as well have less. Thus, we cannot in general say that if a wave function has more nodes than another one it will automatically correspond to a state with higher energy.
Special case: Schroedinger equation in one dimension
There is however a special case:
For the Sturm-Liouville eigenvalue problem (and thus for ordinary second order differential equations with homogeneous boundary conditions) we can strengthen Courant's nodal line theorem such that if we order the possible eigenvalues as $\lambda_1 \leq \lambda_2 \leq \lambda_3 \leq \dots$, then the $n^{\text{th}}$ eigenfunction (the one with energy eigenvalue $\lambda_n$) has exactly $n$ nodes (including the trivial one at the boundary $\mathbf{x} \to \infty$).
This is useful since the one-dimensional time-independent Schrödinger equation is a special case of a Sturm-Liouville equation. So, in the case of the inhomogeneous radial Schrödinger equation with a local potential and node-less inhomogeneity such as the radial Schrödinger equation for the hydrogenic atom
\begin{equation}
\bigg( \frac{ - \hbar^{2} }{ 2 m_{\mathrm{e}} } \frac{ \mathrm{d}^{2} }{ \mathrm{d} r^{2} } + \frac{ \hbar^{2} }{ 2 m_{\mathrm{e}} } \frac{ \ell (\ell + 1) }{ r^{2} } - \frac{ Z e^{2} }{ 2 m_{\mathrm{e}} r } - E \bigg) r R(r) = 0
\end{equation}
it is generally true that a wavefunction with more (radial) nodes must always correspond to a state of higher energy than a wavefunction with less radial nodes. Also, it is clear that the wavefunctions of the one-dimensional particle-in-a-box must follow this rule. But for the three-dimensional particle-in-a-box this is not true anymore, since in that case the Schroedinger equation of the system is not an ordinary second-order differential equation but a partial-differential equation for which only the general version of Courant's nodal line theorem holds.
Some concluding remarks
For real-world system like molecules or crystals the Schroedinger equation is a partial differential equation for which the special case outlined above doesn't apply so that only Courant's nodal line theorem in its general form holds which doesn't give a strict justification for the statement that more nodes mean higher energy. Yet it is very often observed that the number of nodes indeed increases with increasing energy.
The reason for this can be motivated in the following way: The kinetic energy $E_{\mathrm{kin}}$ of a state is proportional to $\int \Psi \Delta \Psi \, d^{3} r$. Via Gauss's theorem it can be shown that $\int \Psi \Delta \Psi \, d^{3} r \propto \int |\nabla \Psi |^{2} \, d^{3} r$ and so $E_{\mathrm{kin}} \propto \int | \nabla \Psi |^{2} d^{3} r$. Now, nodes force a wavefunction to change it's sign. This often means that the value of $\Psi$ has to increase/decrease rather rapidly thus leading to areas with high absolute values of the gradient and thus to high kinetic energy. Since the potential energies shouldn't differ too much between the different states the higher kinetic energy usually also entails a higher total energy. As an example consider the bonding and antibonding wavefunctions of a homonuclear diatomic molecule whose atoms are placed at the positions $r_{\mathrm{A}}$ and $r_{\mathrm{B}}$.
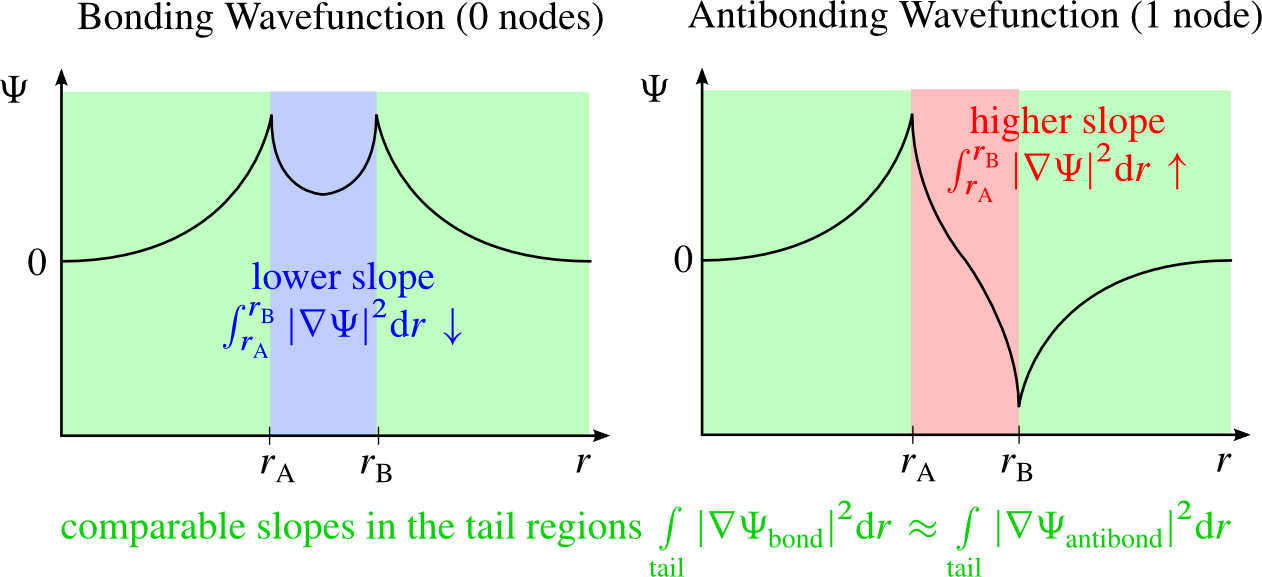
The bonding wavefunction has no nodes. Its value between the atoms doesn't have to undergo a rapid change and thus the slope is rather low. The antibonding wavefunction has one node between the atoms. Its value between the atoms must change rapidly from its positive to its negative maximum thus entailing a very high slope. The slopes of the tail regions are comparable for the bonding and antibonding wavefunctions since it can smoothly fall of to zero at infinity and is not required to go from a maximum value to zero within a very confined region of space - thus even if one wavefunction has to start of at a higher maximum value the gradient will not be much higher. It follows that the antibonding wavefunction has a higher kinetic energy than the bonding wavefunction.
References
[1] R. Courant, D. Hilbert, Methods of Mathematical Physics, Vol. 1, Interscience, New York, 1953, p. 451-455.
[2] R. Courant, "Ein allgemeiner Satz zur Theorie der Eigenfunktionen Selbstadjungierter Differentialausdrücke", Nachr. v. d. Ges. d. Wiss. zu Göttingen 1923, p. 81.