To answer this question it is advantageous to treat a molecule as a graph and use the well known adjacency matrix from graph theory.
Here is the wikipedia definition:
For a simple graph with vertex set $V$, the adjacency matrix is a square $|V| × |V|$ matrix $\mathbb{A}$ such that its element $\mathbb{A}_{ij}$ is one when there is an edge from vertex $i$ to vertex $j$, and zero when there is no edge.
Hückel
The Hückel-Hamiltonian $H$ can be written as:
$$ H = \alpha \mathbf{1} + \beta \mathbb{A}$$
Where $\alpha$ is the ionization energy, $\beta$ the overlap between adjacent $p_z$ orbitals, $\mathbf{1}$ the unit matrix, and $\mathbb{A}$ the adjacency matrix.
If you look at the eigenvalue equation,
$$H \psi = \lambda \psi $$
it has a solution if and only if $\psi$ is an eigenvector of the adjacency matrix.
The eigenvalue $\lambda$ of $H$ is given by the eigenvalue $\tilde{\lambda}$ of the adjacency matrix using the following equation:
$$(\alpha \mathbf{1} + \beta \mathbb{A}) \psi = \alpha \psi + \beta \mathbb{A} \psi = (\alpha + \beta \tilde{\lambda}) \psi$$
What we see is that the $\alpha$ is just a constant offset for the energy. All the relevant information about level spacing, degeneracy etc. is contained in the $\tilde{\lambda}$ with a scaling factor $\beta$.
To put this in another way: All relevant properties of the Hückel-Hamiltionian are encoded in the adjacency matrix of a graph/molecule.
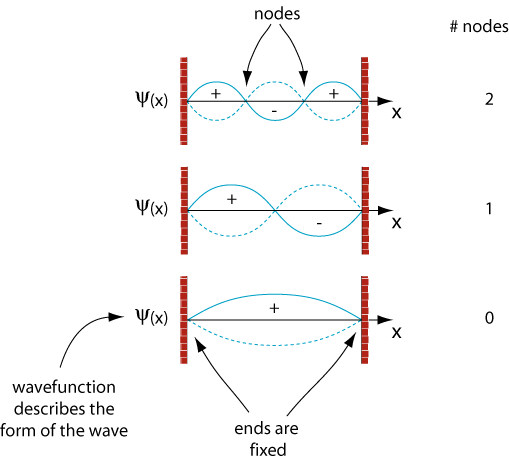
Particle in a box (Free electron network model)
There is a nice paper by Ruedenberg and Scheer 1953 about that topic here.
The main idea is, that your 1D-particle-in-a-box solutions on each edge/bond have to be constrained at joint vertices/atoms.
These constraints are "derived by intuition" in the cited paper.¹
You want the whole wavefunction, that is piecewise composed of the wavefunctions on each edge, to be continous.
Similar to Kirchhoff's Law for electric circuits you want to assert, that the probability current that flows into an vertex/atom flows out.
For this reason, the constraints are called Kirchhoff conditions.
If $V$ is the set of all vertices/atoms, $\psi^e$ labels the wavefunction at the edge/bond $e$, and $E_v$ contains all edges/bonds on a vertex/atom $v$. Then you can express your constraints as:
- Continuity:
$$
\forall v \in V: \forall e_1, e_2 \in E_v: \psi^e_1(v) = \psi^e_2(v)
$$
- Flux Preservation:
$$
\forall v \in V: \sum_{e \in E_v} (\psi^e)' (v) = 0
$$
Without going into the details of the derivation: for cyclic graphs/molecules your eigenvalues $\lambda$ are given again by the eigenvalues $\tilde{\lambda}$ of the adjacency matrix $\mathbb{A}$.
The free scaling factor is then $L$ the edge/bond length (Up to some fixed constants like number literals or $h$.)
Conclusion
For cyclic molecules, it can be proven that the essential properties of the spectrum are given just by the adjacency matrix $\mathbb{A}$ of the molecule in both cases.
The parameter $\alpha$ in the Hückel-Formalism introduces a constant offset, which can be usually ignored for chemistry.
This means that both methods have only one free parameter that scales the spectrum of the adjacency matrix. In the case of Hückel it is the overlap between adjacent $p_z$ orbitals, in the case of the free electron network model it is the bond length $L$.
¹ Note that you can rigorously derive general constraints by forcing only self-adjoint/hermitian realisation of the hamiltonian on the graph. This can be found e.g. here. But we are chemists and not mathematicians so let's stick to the Kirchhoff conditions.