Sorry, but in my opinion this endeavour is unlikely to be successful. The techniques are quite different by their underlying principles. This is, at least in part, why chemists working in (especially, but not limited to) organic sythesis use these and additional ones to get a sufficiently "rounded picture" about the substances analyzed, putting the information each technique provides together.
- mass spectroscopy typically relies on fragmenting the molecules in question, and then separates these then charged fragments by their mass/charge ratio. There are techniques keeping the molecules almost intact (softer ionization techniques, used for example in MS-mapping of tissues), and others (electon impact ionization) where the molecules are intentionally shattered into many differently sized fragments. Depending on the ionization technique applied, the eventually intensity of these individual signals will vary. And if the molecules of interest are embedded in a matrix (e.g., a serum, a tissue), identification of this molecules by MS becomes even more complex.
- NMR spectroscopy typically characterizes probes soluted, keeping the molecules of interest (smaller ones, or large proteins) basically intact, without fragmentation. You basically probe your sample for specific atoms of a kind (isotopes) and their nearest chemical environment. Yet the NMR spectrum of a given sample -- say of an amino acid (building block for proteins) -- already looks differently for scrutinizing the either peripheral hydrogens, the backbone of carbon atoms, or nitrogens.
Addendum
I think a few pictures may provide a glimpse about the complexity here. For a fairly simple molecule like acetophenone, given the molecular structure is known, a sketcher like Chemdoodle may compute within a blink of an eye an estimate how the NMR spectrum either about the hydrogens, or about the carbon atoms will look like. This is possible because the underlying physical principles are known. In addition, there are very many similar compounds analyzed successfully by NMR, allowing (fine tuning) of details in the spectra simulated, e.g. where to expect the signals to appear. Regarding MS, a conservative approach is to predict only the peak(s) that by theory should appear without change of the molecule in terms of its mass. In other words, without fragmentation. Hence Chemdoodle's estimation is this:
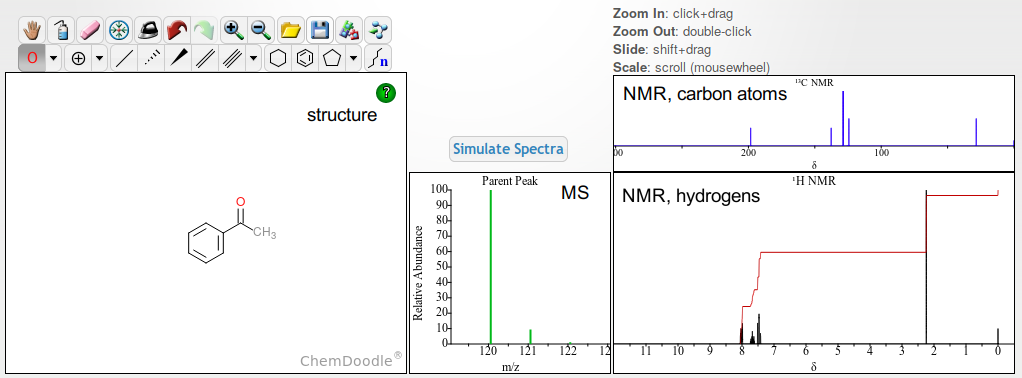 (accessed demo page)
(accessed demo page)
There are equally projects to simulate MS spectra, like CFM-ID. Under the menu Utilities -> Spectra Simulation, you have the possibilty to try out different modi of ionization (shortcut). Acetophenone's structure, expressed in a machine-readable Smiles code is
C1=CC=C(C=C1)C(=O)C
Then, just choosing ESI mode about positive ions, without considering adducts, the three MS spectra simulated by CFM-ID of actephenone, in increasing hardness of ionization are these:
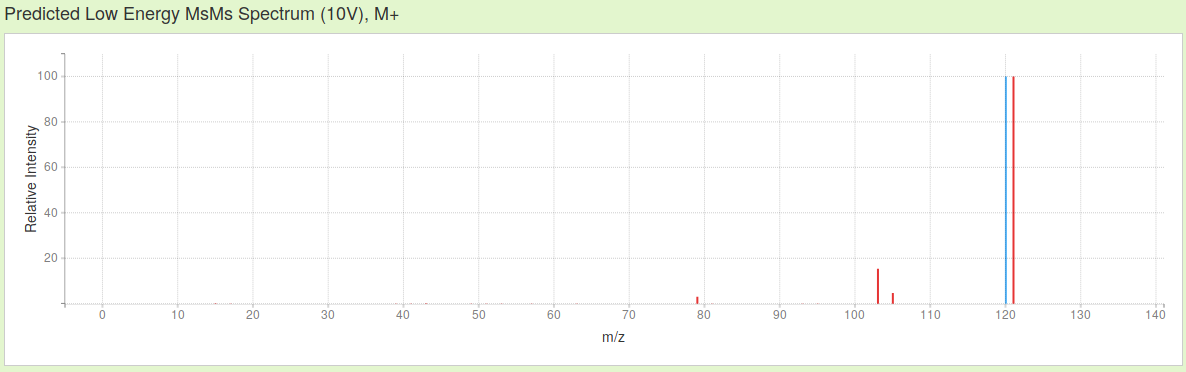
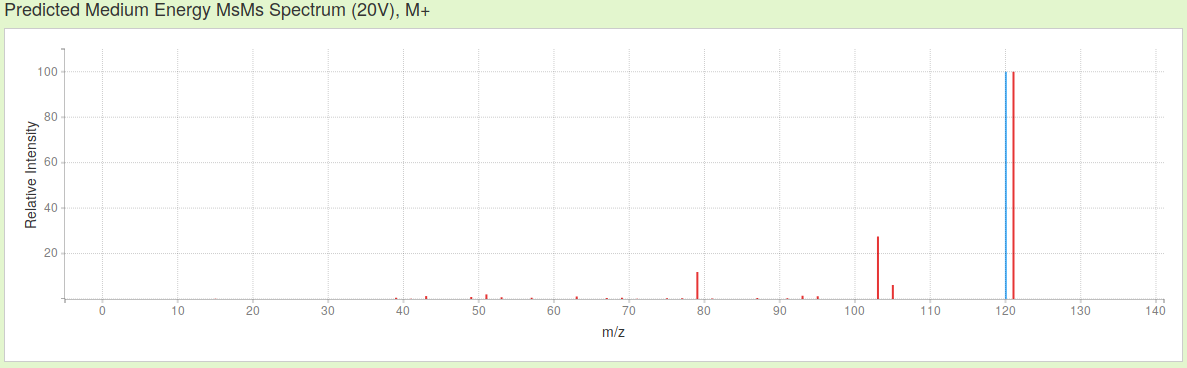
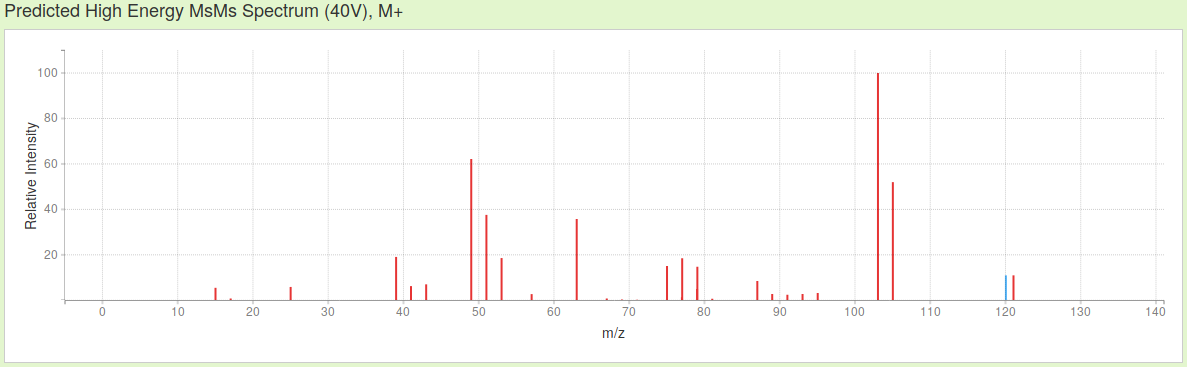
As you see, not only the presence / absence of the signals changes in function of the probability to record certain fragments of a specific mass/charge ratio (m/z), their relative intensity equally is affected by changing the parameters of ionization.
(Hovering the mouse over the streaks allows you to display the suggested fragments responsible for the signals in these simulations. Because of the special conditions in a mass spectrometer, unstable specis you wouldn't be able to store in a shelf may be seen, too.)