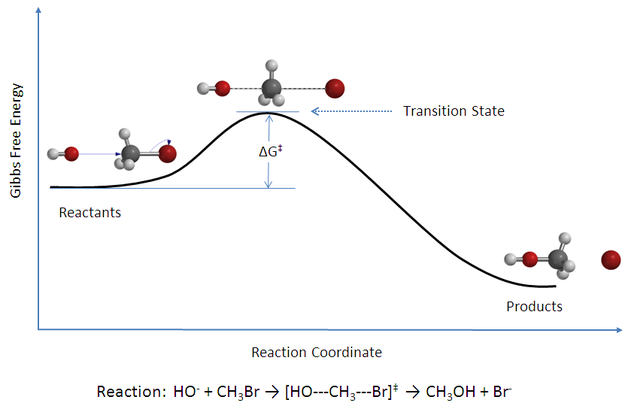
You are correct in your analysis that the rate constant is first order with units $\ce s^{-1}$. The problem you have is that the Eyring equation you use is not complete as I try to show below after some introduction. (The derivation I use can be found in several texts, but I outline that in Steinfeld, Fransisco & Hase, 'Chemical Kinetics & Dynamics')
The idea behind Transition State Theory (TST) is that the reactants are assumed to be in thermal equilibrium with the transition state. This is a small region at the top of the potential energy barrier that separates reactants and products. A contour plot of an A + BC reaction ($\ce{F + H2}$) is shown in the first figure (together with a single reaction trajectory).
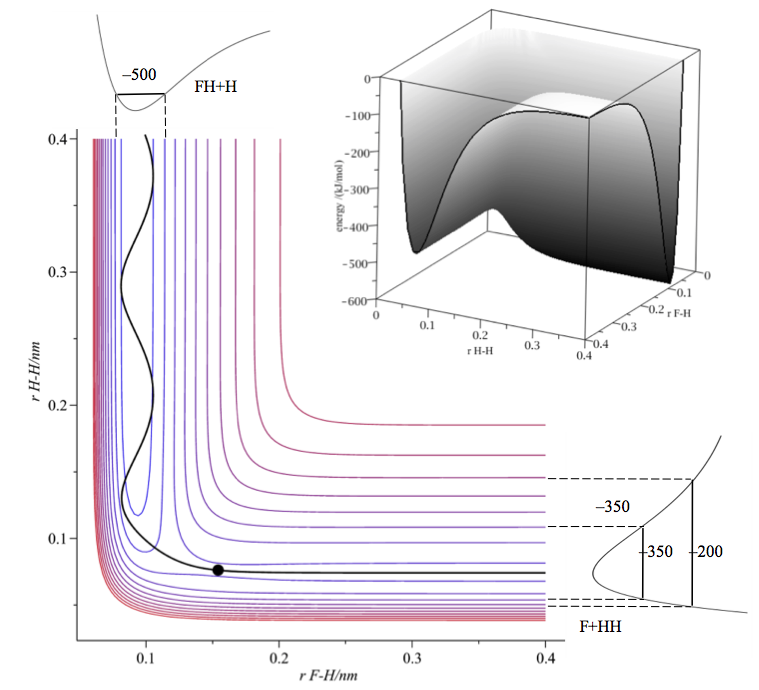
The transition state is labelled with a dot and lies at a saddle point, which is to say that the potential is convex along the reaction path and concave in a perpendicular direction to this, see second figure.
Following the reaction path from right to left (contour first figure) we see that on collision the reactants climb the (rather small) barrier and cross to products. The product is formed with some considerable vibrational energy. The profile along the reaction path (or reaction coordinate) is the familiar potential energy hill between reactants and products.
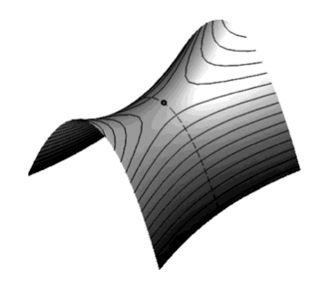
There are a number of assumptions in deriving TST. The two basic ones are
(a) that nuclear and electron motion can each be separated (similarly to the Born-Oppenheimer approximation use to solve the Schrodinger equation), and it is further assumed that the energy for translation, vibration and rotational motion can be treated additively),
(b) the reactant molecule’s energy is distributed among its states, i.e. translation, vibration, rotation according to the Maxwell-Boltzmann distribution.
Other assumptions are necessary for the theory and are that:
(c) the transition state is crossed only once, i.e. it is a point of no return,
(d) The motion along the reaction coordinate at the transition state can be separated from other motions and treated classically as a translation.
(e) even in the absence of equilibrium between reactants and product molecules the energy in the modes of the transition state is distributed according to Maxwell-Boltzmann. This assumption is sometimes called the ‘quasi-equilibrium hypothesis’.
To calculate the rate constant $k$, the speed of crossing the transition state and the number of molecules doing so has to be calculated. Let the reaction be a general second order reaction - $\ce{A + B -> C}$ with rate equation:
$$\frac{\mathrm d[A]}{\mathrm dt}=-k\ce{[A][B]}$$
In the reaction $\ce{A + B -> X^\ddagger -> C}$, where $\ce{X^\ddagger}$ is the transition state, suppose that there is a small region of length $\delta$ at the top of the barrier that the reactants must all pass through.
Imagine this as a very thin dividing surface, reactants on one side and products on the other. At equilibrium the rate of forwards and backwards motion through this region must be the same, consequently if the concentration is $N^\ddagger$ then:
$$N^\ddagger = K^\ddagger\ce{[A][B]}$$
Now suppose that all the products are removed from equilibrium and as these are half of the total $N^\ddagger$ the number going forwards to products
$$N_\mathrm f^\ddagger = K^\ddagger\frac{[A][B]}{2}$$
which is the quasi equilibrium hypothesis.
The rate of reaction to reactants to products is given as:
$$\frac{\mathrm dN}{\mathrm dt} = \frac{\delta _{N^\ddagger}}{\delta_t}$$
Here, the number of transition states with velocity $v$ to $v+\delta v$ in one direction is:
$$\delta_{N^\ddagger} = \frac{N^\ddagger}{2}$$
The average time to cross the dividing surface at average velocity $v_\mathrm a$ is:
$$\delta_t = \frac{\delta}{v_\mathrm a}$$
where $\delta$ is the thickness of the dividing surface and by substituting gives
$$\frac{\mathrm dN}{\mathrm dt} = \frac{N^\ddagger}{2}\cdot\frac{v_\mathrm a }{ \delta }$$
The average velocity in one direction can be calculated in the standard way$^1$ assuming equilibrium distribution of velocities and is:
$$v_\mathrm a = \left(\frac{2k_\mathrm BT}{\pi\mu}\right)^{1/2}$$
where $\mu$ is the reduced mass. Substituting again gives:
$$\frac{\mathrm dN}{\mathrm dt}=\frac{N^\ddagger}{2\delta}\cdot\left(\frac{2k_\mathrm BT}{\pi\mu}\right)^{1/2}$$
The equilibrium constant is given by:
$$K^\ddagger= \frac{N^\ddagger}{\ce{[A][B]}} = \frac{Z_\mathrm{tot}^\ddagger}{Z_\mathrm A Z_\mathrm B}e^{-E_0/k_\mathrm BT} $$
Where $Z_\mathrm{tot}^\ddagger$, $Z_\mathrm A$ and $Z_\mathrm B$ are the respective partition functions per unit volume and are derived using statistical mechanics.
The $Z_\mathrm {tot}^\ddagger$ partition function is that of the combined species $\ce{AB}$ at the transition state which by the condition (d) above, is separated into two terms - one term for the reaction coordinate($Z_\mathrm r$) and $Z^\ddagger$ for all other $3N-1$ degrees of freedom.
$$Z_\mathrm{tot}^\ddagger = Z_rZ^\ddagger$$
The translational partition function in one dimension is
$$Z_r=\left(2\pi\mu k_BT\right)^{1/2}\cdot \frac{\delta}{h}$$
Thus substituting and simplifying produces:
$$\frac{\mathrm dN}{\mathrm dt}= \frac{k_\mathrm BT}{h}\frac{Q^\ddagger}{Q_\mathrm AQ_\mathrm B}e^{-E_0/k_\mathrm BT}\ce{[A][B]}$$
from which we get the second order rate constant to be:
$$k= \frac{k_BT}{h}\frac{Q^\ddagger}{Q_\mathrm AQ_\mathrm B}e^{-E_0/k_\mathrm BT}$$
The constants $\delta$ and $\mu$, which were used in the derivation, cancel out. This constant $(k_\mathrm BT/h = \pu{6.25 \times 10^{12} s-1})$ is often called the frequency factor.
The rate constant will have units of $\pu {m3 s-1}$
All that now remains is to calculate the partition functions.
The parameters needed have to be guessed for the transition state as they are generally unknown, no transition state ever having being identified spectroscopically. The values needed are vibrational frequencies, moments of inertia reactants and shape (linear or bent). The general partition function is split into a product of terms, $Z_\mathrm {trans} \cdot Z_\mathrm {rot} \cdot Z_\mathrm {vib} \cdot Z_\mathrm {elec}$. This is assumption (a) above, separating electronic and nuclear motions.
In the case of atom plus diatom reaction $\ce{A + BC -> AB + C}$ of which many have been measured - $\ce{O + H2, Cl + H2, H + H2, F + H2}$ etc. - the rate constant equation becomes the monster equation:
$$k=\frac{k_\mathrm BT}{h}\left[\frac{Z^\ddagger}{Z_\mathrm A Z_\mathrm {BC}}\right]_\mathrm {vib}\left[\frac{Z^\ddagger}{Z_\mathrm A Z_\mathrm {BC}}\right]_\mathrm {rot}\left[\frac{Z^\ddagger/V}{Z_\mathrm A/V \cdot Z_\mathrm {BC}/V}\right]_\mathrm {trans}e^{-E_0/k_\mathrm BT}$$
Finally, the whole rate constant may need to be multiplied by a symmetry or statistical factor, for example - in $\ce{F + H2}$ the rate constant needs an addition factor of $2$.
The rate constant has units $\pu{m3 s-1}$ which when multiplied by the Avogadro number becomes $\pu{dm3 mol-1 s-1}$, normal $2^\mathrm {nd}$ order units.
Notes:
(A) The volume appears because we need to convert the thermodynamic equilibrium constant $k$ (ratio of partition functions) into $k_c$, the chemical one which uses concentrations.
(B) The equations for partition functions are well known, are listed on many phys. chem. textbooks, e.g. McQuarrie & Simon ‘Physical Chemistry’ chapter 1, but the Wikipedia page is rather technical so they are listed below$^{2}$.
(C) It is important to note that partition functions have widely differing values, translation $\frac{Z_\mathrm{trans}^\ddagger}{V} \approx \pu{10^{11}m-3}$; rotation $Z_\mathrm{rot}^\ddagger \approx 10^4 $, vibration $Z_\mathrm{vib}^\ddagger \approx 10$ and electronic $Z_\mathrm{elec}^\ddagger \approx 1$ are all dimensionless. (Thus, the translational partition function divided by $V$ has dimensions of $\pu{m-3}$).
Limitations:
(A)As the transition state properties are not known, reactions between complicated molecules are unlikely to give accurate values.
(B) Molecules in solution are limited by diffusion to a maximum value determined by diffusion constants and hence solution viscosity.
(C) More recent treatments by Kramers discuss how, in solution, the rate constant depends on friction of the reaction coordinate (see Daune ‘Molecular Biophysics).
Footnotes
(1) The average of a quantity $x$ with distribution $p(x)$ is calculated with the integral ratio
$$\frac{\int \exp(x)dx}{\int p(x)dx}$$
(The denominator is the normalizing term, equivalent to the total number when making a simple average of any set of objects, i.e. $\sum (x/N)$)
In the case in point, the average velocity distribution is $p(v) = \exp(-\mu v^2/(2k_BT))$ and the integration limits are 0 to $\infty$.
(2) The partition function is defined as the ‘sum over states’,
$$Z=\sum_i g_i \exp(-E_i/(k_\mathrm BT))$$
where $g_i$ is the degeneracy of the energy levels $E_i$.
When the energy levels are close (as in translation) the summation is replaced with an integral.
The partition functions are
$$Z_\mathrm {trans} = \frac{(2\pi \mu k_\mathrm BT)^{3/2}V}{h^3}$$
For a generally shaped molecule, with moments of inertia $I_\mathrm a$ etc. about the three principal axes, and symmetry factor $\sigma$ (which is the number of ways the molecule can be rotated back to the original):
$$Z_\mathrm {rot} = \left(\frac{8\pi ^2k_BT}{h^2}\right)^{3/2}\cdot\frac{\left(\pi I_aI_bI_c\right)^{1/2}}{\sigma}$$
For each vibrational mode.
$$Z_\mathrm {vib} = \frac{1}{1-e^{-h\nu/k_\mathrm BT}}$$
These partition functions are multiplied together for many vibrational modes; $\nu$ is the vibrational frequency.
The electronic partition function is the multiplicity of the state, most commonly 1.