Even though this article does not use the Valence Bond Approach, one can clearly see the qualitative manifestation of Bent's Rule (compare also: Utility of Bent's Rule - What can Bent's rule explain that other qualitative considerations cannot?Utility of Bent's Rule - What can Bent's rule explain that other qualitative considerations cannot?). A higher $s$ character means a shorter bond. The lack of reactivity towards nucleophiles can be explained electronically with a delocalised LUMO. In terms of localised bonding, the lone pairs of any additional chlorine atom would provide sufficient electron density, to shield the backside attack on the carbon.
replaced http://chemistry.stackexchange.com/ with https://chemistry.stackexchange.com/
#Introduction (and Abstract, TLDR) In very short words you can say, that the anomeric effect is responsible for the lack of reactiveness. The electronic effect may very well be compensating for the the steric effect, that could come from the methyl moiety. In any way, most of the steric effects can often been seen as electronic effects in disguise.
#Analysis of Molecular Orbitals I will analyse the bonding picture based on calculations at the density fitted density functional level of theory, with a fairly large basis set: DF-BP86/def2-TZVPP. As model compounds I have chosen chloromethane, dichloromethane, chloroform and chloroethane.
First of all let me state, that the bond lengths are a little larger at this level, however, the general trend for shortening can also be observed. In this sense, chloroethane behaves like chloromethane. An attempt to explain this will be given at the end of this article.
\begin{array}{lr}\hline \text{Compound} & \mathbf{d}(\ce{C-Cl})\\\hline \ce{ClCH3} & 1.797\\ \ce{Cl2CH2} & 1.786\\ \ce{Cl3CH} & 1.783\\\hline \ce{ClCH2CH3} & 1.797\\\hline \end{array}
In the canonical bonding picture, it is fairly obvious, that the electronic effects dominate and are responsible for the lack of reactivity. In other words, the lowest unoccupied molecular orbital is very well delocalised in the dichloromethane and chloroform case. This is effectively leaving no angle to attack the antibonding orbitals.
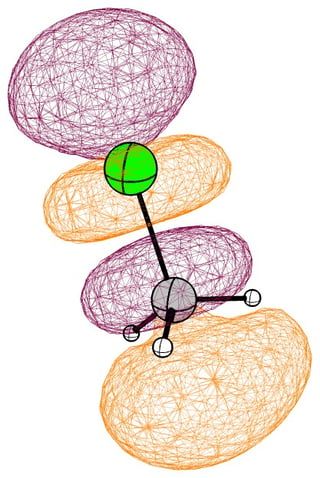
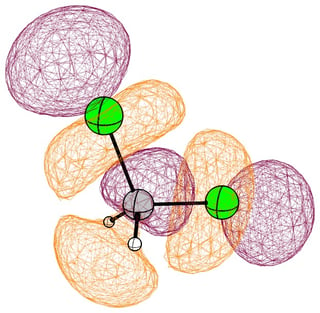
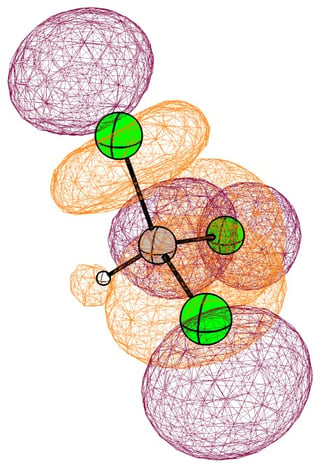
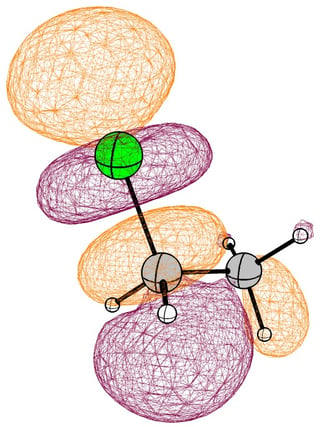
In the mono substituted cases there is a large coefficient at the carbon, where a nucleophile can readily attack.
One can also analyse the bonding situation in terms of localised orbitals. Here I make use of a Natural Bond Orbital (NBO) analysis, that transforms the canonical orbitals into hybrid orbitals, which all have an occupation of about two electrons. Due to the nature of the approach, it is no longer possible to speak of HOMO or LUMO, when analysing the orbitals. Due to the nature of the calculations, i.e. there are polarisation functions, the values do not necessarily add up to 100%. The deviation is so small, that it can be omitted.
The following table shows the composition (in $\%$)of the carbon chloro bond and anti bond.
\begin{array}{lrr}\hline
\text{Compound} &\sigma-\ce{C-Cl} & \sigma^*-\ce{C-Cl}\\\hline
\ce{ClCH3} & 45\ce{C}(21s79p) 55\ce{Cl}(14s85p)
& 55\ce{C}(21s79p) 45\ce{Cl}(14s85p)\\
\ce{Cl2CH2} & 46\ce{C}(22s77p) 54\ce{Cl}(14s85p)
& 54\ce{C}(22s77p) 46\ce{Cl}(14s85p)\\
\ce{Cl3CH} & 48\ce{C}(24s76p) 52\ce{Cl}(14s86p)
& 52\ce{C}(24s76p) 48\ce{Cl}(14s86p)\\\hline
\ce{ClCH2CH3} & 44\ce{C}(19s81p) 56\ce{Cl}(14s85p)
& 56\ce{C}(19s81p) 44\ce{Cl}(14s85p)\\\hline
\end{array}
As we go from mono- to di- to trisubstituted methane, the carbon contribution increases slightly, along with the percentage of $s$ character. More $s$ character usually means also a stronger bond, which often results in a shorter bond distance. Of course, delocalization will have a similar effect on its own.
The reason, why dichloromethane and chloroform are fairly unreactive versus nucleophiles, has already been pointed out in terms of localised bonding. But we can have a look at these orbitals as well.
In the case of chloromethane, the LUMO has more or less the same scope of the canonical orbital, with the highest contribution from the carbon. If we compare this antibonding orbital to an analogous orbital in dichloromethane or chloroform, we can expect the same form. We soon run into trouble, because of the localised $p$ lone pairs of chlorine. Not necessarily overlapping, but certainly in the way of the "backside" of the bonding orbital. In the case of chloroethane we can observe hyperconjugation. However, this effect is probably less strong, and from the canonical bonding picture we could also assume, that this increases the polarisation of the antibonding orbital in favour of carbon.
In the following pictures, occupied orbitals are coloured red and yellow, while virtual orbitals are coloured purple and orange.
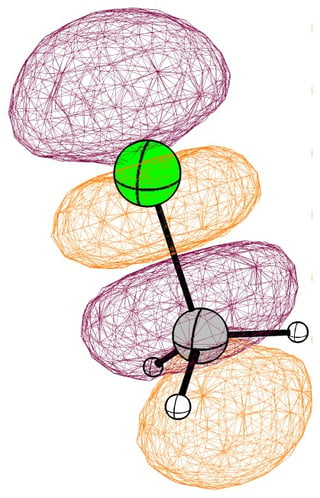
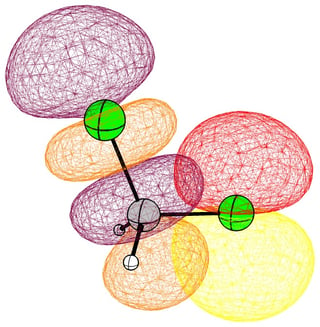
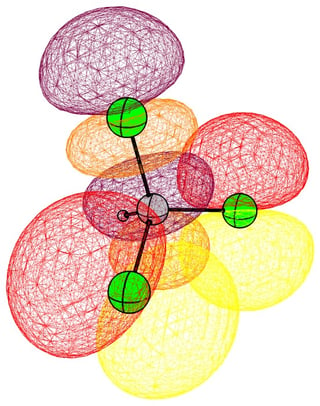
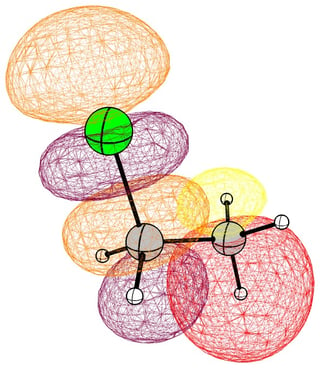
(Note that in chloroform two lone pair orbitals are shown.)
#Conclusion
Even though this article does not use the Valence Bond Approach, one can clearly see the qualitative manifestation of Bent's Rule (compare also: Utility of Bent's Rule - What can Bent's rule explain that other qualitative considerations cannot?). A higher $s$ character means a shorter bond. The lack of reactivity towards nucleophiles can be explained electronically with a delocalised LUMO. In terms of localised bonding, the lone pairs of any additional chlorine atom would provide sufficient electron density, to shield the backside attack on the carbon.